[1]
Crist E, Mora C, Engelman R. The interaction of human population, food production, and biodiversity protection[J]. Science, 2017, 356(6335): 260⁃264.
[本文引用: 1]
[2]
Güney T. Renewable energy, non⁃renewable energy and sustainable development[J]. International Journal of Sustainable Development & World Ecology, 2019, 26(5): 389⁃397.
[本文引用: 1]
[3]
Lin L F, Han X, Han B X, et al. Emerging heterogeneous catalysts for biomass conversion: Studies of the reaction mechanism[J]. Chemical Society Reviews, 2021, 50: 11270⁃11292.
[本文引用: 2]
[4]
Zhang T. Taking on all of the biomass for conversion[J]. Science, 2020, 367(6484): 1305⁃1306.
[本文引用: 1]
[5]
Blanco E, Aguirre A D A, de León J N D, et al. Relevant aspects of the conversion of guaiacol as a model compound for bio⁃oil over supported molybdenum oxycarbide catalysts[J]. New Journal of Chemistry, 2020, 44(28): 12027⁃12035.
[本文引用: 4]
[6]
Sangnikul P, Phanpa C, Xiao R, et al. Role of copper⁃or cerium⁃promoters on NiMo/γ⁃Al2 O3 catalysts in hydrodeoxygenation of guaiacol and bio⁃oil[J]. Applied Catalysis A: General, 2019, 574: 151⁃160.
[本文引用: 2]
[7]
Li T, Li H, Huang G M, et al. Transforming biomass tar into a highly active Ni⁃based carbon⁃supported catalyst for selective hydrogenation⁃transalkylation of guaiacol[J]. Journal of Analytical and Applied Pyrolysis, 2020, 153: 104976⁃104983.
[本文引用: 5]
[8]
Zhou M H, Ye J, Liu P, et al. Water⁃assisted selective hydrodeoxygenation of guaiacol to cyclohexanol over supported Ni and Co bimetallic catalysts[J]. ACS Sustainable Chemistry & Engineering, 2017, 5(10): 8824⁃8835.
[本文引用: 10]
[9]
Adilina I B, Rinaldi N, Simanungkalit S P, et al. Hydrodeoxygenation of guaiacol as a bio⁃oil model compound over pillared clay⁃supported nickel⁃molybdenum catalysts[J]. The Journal of Physical Chemistry C, 2019, 123: 21429⁃21439.
[本文引用: 6]
[10]
Lai Q H, Zhang C, Holles J H. Hydrodeoxygenation of guaiacol over Ni@Pd and Ni@Pt bimetallic overlayer catalysts[J]. Applied Catalysis A: General, 2016, 528: 1⁃13.
[本文引用: 4]
[11]
Gutiérrez⁃Rubio S, Berenguer A, Přech J, et al. Guaiacol hydrodeoxygenation over Ni2 P supported on 2D⁃zeolites[J]. Catalysis Today, 2020, 345: 48⁃58.
[本文引用: 5]
[12]
Zhang X Q, Yan P F, Zhao B, et al. Identification of electron⁃rich mononuclear Ni atoms on TiO2 ⁃A distinguished from Ni particles on TiO2 ⁃R in guaiacol hydrodeoxygenation pathways[J]. Catalysis Science & Technology, 2021, 11(1): 297⁃311.
[本文引用: 5]
[13]
Jin W, Pastor P L, Villora⁃Pico J J, et al. In⁃situ HDO of guaiacol over nitrogen⁃doped activated carbon supported nickel nanoparticles[J]. Applied Catalysis A: General, 2021, 620: 118033⁃118040.
[本文引用: 1]
[14]
Rong Z, Lu J Q, Yu G Q, et al. Promoting selective hydrodeoxygenation of guaiacol over amorphous nanoporous NiMnO2 [J]. Catalysis Communications, 2020, 140: 105987⁃105991.
[本文引用: 3]
[15]
Lu M H, Jiang Y J, Sun Y, et al. Hydrodeoxygenation of guaiacol catalyzed by ZrO2 ⁃CeO2 supported nickel catalysts with high⁃loading[J]. Energy & Fuels, 2020, 34: 4685⁃4692.
[本文引用: 2]
[16]
Zhang X Q, Yan P F, Zhao B, et al. Selective hydrodeoxygenation of guaiacol to phenolics by Ni/anatase TiO2 catalyst formed by cross⁃surface migration of Ni and TiO2 [J]. ACS Catalysis, 2019, 9(4): 3551⁃3563.
[本文引用: 4]
[17]
Lu M H, Sun Y, Zhang P, et al. Hydrodeoxygenation of guaiacol catalyzed by high⁃loading Ni catalysts supported on SiO2 ⁃TiO2 binary oxides[J]. Industrial & Engineering Chemistry Research, 2019, 58(4): 1513⁃1524.
[本文引用: 3]
[18]
Lu J Q, Liu X, Yu G Q, et al. Selective hydrodeoxygenation of guaiacol to cyclohexanol catalyzed by nanoporous nickel[J]. Catalysis Letters, 2020, 150(3): 837⁃848.
[本文引用: 2]
[19]
Fan X D, Wu Y J, Tu R, et al. Hydrodeoxygenation of guaiacol via rice husk char supported Ni based catalysts: The influence of char supports[J]. Renewable Energy, 2020, 157: 1035⁃1045.
[本文引用: 3]
[20]
Lan X F, Hensen E J M, Weber T, et al. Hydrodeoxygenation of guaiacol over Ni2 P/SiO2 ⁃reaction mechanism and catalyst deactivation[J]. Applied Catalysis A: General, 2018, 550: 57⁃66.
[本文引用: 5]
[21]
Tran Q K, Ly H V, Kwon B, et al. Catalytic hydrodeoxygenation of guaiacol as a model compound of woody bio⁃oil over Fe/AC and Ni/γ⁃Al2 O3 catalysts[J]. Renewable Energy, 2021, 173: 886⁃895.
[本文引用: 3]
[22]
Wang H H, Liu Z L, Song Y C, et al. High⁃performance evolution of Ni2 P@hierarchical HZSM⁃5 as the guaiacol hydrodeoxygenation catalyst[J]. ACS Omega, 2020, 5(34): 21330⁃21337.
[本文引用: 4]
[23]
Liu L J, Liu Y G, Gao X, et al. Hydrodeoxygenation of bio⁃oil model compounds over amorphous NiB/SiO2 ⁃Al2 O3 catalyst in oil⁃water biohasic system[J]. Journal of Fuel Chemistry & Technology, 2017, 45(8): 932⁃938.
[本文引用: 5]
[24]
Blanco E, Dongil A B, Escalona N. Synergy between Ni and Co nanoparticles supported on carbon in guaiacol conversion[J]. Nanomaterials (Basel, Switzerland), 2020, 10(11): 2199⁃2216.
[本文引用: 6]
[25]
Chen C Z, Zhou M H, Liu P, et al. Flexible NiCo⁃based catalyst for direct hydrodeoxygenation of guaiacol to cyclohexanol[J]. New Journal of Chemistry, 2020, 44(43): 18906⁃18916.
[本文引用: 7]
[26]
Patil M L, Lali A M, Dalai A K. Catalytic hydrodeoxygenation of bio⁃oil model compound for production of fuel grade oil[J]. Asia⁃Pacific Journal of Chemical Engineering, 2019, 14(4): 2317⁃2330.
[本文引用: 5]
[27]
Tran C C, Stankovikj F, Garcia⁃Perez M, et al. Unsupported transition metal⁃catalyzed hydrodeoxygenation of guaiacol[J]. Catalysis Communications, 2017, 101: 71⁃76.
[本文引用: 2]
[28]
He T, Liu X X, Ge Y Z, et al. Gas phase hydrodeoxygenation of anisole and guaiacol to aromatics with a high selectivity over Ni⁃Mo/SiO2 [J]. Catalysis Communications, 2017, 102: 127⁃130.
[本文引用: 3]
[29]
Lima R W S, Hewer T L R, Alves R M B, et al. Surface analyses of adsorbed and deposited species on the Ni⁃Mo catalysts surfaces after Guaiacol HDO. Influence of the alumina and SBA⁃15 supports[J]. Molecular Catalysis, 2021, 511: 111724⁃111733.
[本文引用: 2]
[30]
Mukundan S, Wahab M A, Atanda L, et al. Highly active and robust Ni⁃MoS2 supported on mesoporous carbon: A nanocatalyst for hydrodeoxygenation reactions[J]. RSC Advances, 2019, 9(30): 17194⁃17202.
[本文引用: 5]
[31]
Chen Q, Cai C L, Zhang X H, et al. Amorphous FeNi⁃ZrO2 ⁃catalyzed hydrodeoxygenation of lignin⁃derived phenolic compounds to naphthenic fuel[J]. ACS Sustainable Chemistry & Engineering, 2020, 8: 9335⁃9345.
[本文引用: 3]
[32]
Fang H H, Zheng J W, Luo X L, et al. Product tunable behavior of carbon nanotubes⁃supported Ni⁃Fe catalysts for guaiacol hydrodeoxygenation[J]. Applied Catalysis A: General, 2017, 529: 20⁃31.
[本文引用: 9]
[33]
Hamid S B A, Ambursa M M, Sudarsanam P, et al. Effect of Ti loading on structure⁃activity properties of Cu⁃Ni/Ti⁃MCM⁃41 catalysts in hydrodeoxygenation of guaiacol[J]. Catalysis Communications, 2017, 94: 18⁃22.
[本文引用: 5]
[34]
Niu X X, Wang L W, Chen J X. Improved performance of SiO2 ⁃supported Ni3 Ga intermetallic compound for deoxygenation of phenolic compounds[J]. Catalysis Communications, 2020, 140: 106001⁃106006.
[本文引用: 4]
[35]
Park C W, Kim J W, Kim H U, et al. Bimetallic Ni‐Re catalysts for the efficient hydrodeoxygenation of biomass⁃derived phenols[J]. International Journal of Energy Research, 2021, 45(11): 16349⁃16361.
[本文引用: 4]
[36]
Sirous⁃Rezaei P, Jae J, Cho K, et al. Insight into the effect of metal and support for mild hydrodeoxygenation of lignin⁃derived phenolics to BTX aromatics[J]. Chemical Engineering Journal, 2019, 377: 120121.
[本文引用: 3]
[37]
López M, Palacio R, Royer S, et al. Mesostructured CMK⁃3 carbon supported Ni⁃ZrO2 as catalysts for the hydrodeoxygenation of guaiacol[J]. Microporous and Mesoporous Materials, 2020, 292: 109694.
[本文引用: 3]
[38]
Parrilla⁃Lahoz S, Jin W, Pastor⁃Pérez L, et al. Guaiacol hydrodeoxygenation in hydrothermal conditions using N⁃doped reduced graphene oxide (RGO) supported Pt and Ni catalysts: Seeking for economically viable biomass upgrading alternatives[J]. Applied Catalysis A: General, 2021, 611: 117977⁃117986.
[本文引用: 2]
[39]
García⁃Mendoza C, Santolalla⁃Vargas C E, Woolfolk L G, et al. Effect of TiO2 in supported NiWS catalysts for the hydrodeoxygenation of guaiacol[J]. Catalysis Today, 2020, 377: 145⁃156.
[本文引用: 3]
[40]
Ansaloni S, Russo N, Pirone R. Hydrodeoxygenation of guaiacol over molybdenum‐based catalysts: The effect of support and the nature of the active site[J]. The Canadian Journal of Chemical Engineering, 2017, 95(9): 1730⁃1744.
[本文引用: 4]
[41]
Moreira R, Ochoa E, Pinilla J L, et al. Liquid⁃phase hydrodeoxygenation of guaiacol over Mo2 C supported on commercial CNF. Effects of operating conditions on conversion and product selectivity[J]. Catalysts, 2018, 8(4): 127.
[本文引用: 2]
[42]
Ochoa E, Torres D, Pinilla J L, et al. Nanostructured carbon material effect on the synthesis of carbon⁃supported molybdenum carbide catalysts for guaiacol hydrodeoxygenation[J]. Energies, 2020, 13(5): 1189.
[本文引用: 1]
[43]
Ochoa E, Torres D, Moreira R, et al. Carbon nanofiber supported Mo2 C catalysts for hydrodeoxygenation of guaiacol: The importance of the carburization process[J]. Applied Catalysis B: Environmental, 2018, 239: 463⁃474.
[本文引用: 2]
[44]
Tran C C, Han Y L, Garcia⁃Perez M, et al. Synergistic effect of Mo⁃W carbides on selective hydrodeoxygenation of guaiacol to oxygen⁃free aromatic hydrocarbons[J]. Catalysis Science & Technology, 2019, 9(6): 1387⁃1397.
[本文引用: 1]
[45]
Cai Z, Wang F M, Zhang X B, et al. Selective hydrodeoxygenation of guaiacol to phenolics over activated carbon supported molybdenum catalysts[J]. Molecular Catalysis, 2017, 441: 28⁃34.
[本文引用: 4]
[46]
Han G H, Lee M W, Park S, et al. Revealing the factors determining the selectivity of guaiacol HDO reaction pathways using ZrP⁃supported Co and Ni catalysts[J]. Journal of Catalysis, 2019, 377: 343⁃357.
[本文引用: 2]
[47]
Mei X, Wu D F. Highly selective catalytic conversion of lignin⁃derived phenolic compounds to cycloalkanes over a hierarchically structured zeolite catalyst[J]. Journal of Materials Science, 2019, 54(4): 2940⁃2959.
[本文引用: 2]
[48]
Liu G H, Zong Z M, Liu Z Q, et al. Solvent⁃controlled selective hydrodeoxygenation of bio⁃derived guaiacol to arenes or phenols over a biochar supported Co⁃doped MoO2 catalyst[J]. Fuel Processing Technology, 2018, 179: 114⁃123.
[本文引用: 4]
[49]
Gutierrez A, Turpeinen E M, Viljava T R, et al. Hydrodeoxygenation of model compounds on sulfided CoMo/γ⁃Al2 O3 and NiMo/γ⁃Al2 O3 catalysts; role of sulfur⁃containing groups in reaction networks[J]. Catalysis Today, 2017, 285: 125⁃134.
[本文引用: 1]
[50]
Mora⁃Vergara I D, Moscoso L H, Gaigneaux E M, et al. Hydrodeoxygenation of guaiacol using NiMo and CoMo catalysts supported on alumina modified with potassium[J]. Catalysis Today, 2018, 302: 125⁃135.
[本文引用: 2]
[51]
Li C C, Nakagawa Y, Tamura M, et al. Hydrodeoxygenation of guaiacol to phenol over ceria⁃supported iron catalysts[J]. ACS Catalysis, 2020, 10(24): 14624⁃14639.
[本文引用: 2]
[52]
Olcese R N, Bettahar M, Petitjean D, et al. Gas⁃phase hydrodeoxygenation of guaiacol over Fe/SiO2 catalyst[J]. Applied Catalysis B, Environmental, 2012, 115⁃116: 63⁃73.
[本文引用: 2]
[53]
Tran N T T, Uemura Y, Ramli A, et al. Vapor⁃phase hydrodeoxygenation of lignin⁃derived bio⁃oil over Al⁃MCM⁃41 supported Pd⁃Co and Pd⁃Fe catalysts[J]. Molecular Catalysis, 2021,523: 111435⁃111445.
[本文引用: 5]
[54]
Tran N, Uemura Y, Trinh T, et al. Hydrodeoxygenation of guaiacol over Pd⁃Co and Pd⁃Fe catalysts: Deactivation and regeneration[J]. Processes, 2021, 9(3): 430.
[本文引用: 3]
[55]
Wang X C, Wu P X, Wang Z Q, et al. Chlorine⁃modified Ru/TiO2 catalyst for selective guaiacol hydrodeoxygenation[J]. ACS Sustainable Chemistry & Engineering, 2021, 9(8): 3083⁃3094.
[本文引用: 6]
[56]
Dwiatmoko A A, Kim I, Zhou L, et al. Hydrodeoxygenation of guaiacol on tungstated zirconia supported Ru catalysts[J]. Applied Catalysis A: General, 2017, 543: 10⁃16.
[本文引用: 1]
[57]
Lin B Q, Li R X, Shu R Y, et al. Synergistic effect of highly dispersed Ru and moderate acid site on the hydrodeoxygenation of phenolic compounds and raw bio⁃oil[J]. Journal of the Energy Institute, 2020, 93(3): 847⁃856.
[本文引用: 2]
[58]
Roldugina E A, Naranov E R, Maximov A L, et al. Hydrodeoxygenation of guaiacol as a model compound of bio⁃oil in methanol over mesoporous noble metal catalysts[J]. Applied Catalysis A: General, 2018, 553: 24⁃35.
[本文引用: 2]
[59]
Yan P H, Mensah J, Drewery M, et al. Role of metal support during ru⁃catalysed hydrodeoxygenation of biocrude oil[J]. Applied Catalysis B: Environmental, 2021, 281: 119470⁃119481.
[本文引用: 2]
[60]
Hu L, Wei X Y, Zong Z M. Ru/Hβ catalyst prepared by the deposition⁃precipitation method for enhancing hydrodeoxygenation ability of guaiacol and lignin⁃derived bio⁃oil to produce hydrocarbons[J]. Journal of the Energy Institute, 2021, 97(7): 48⁃57.
[本文引用: 2]
[61]
Kim M, Ha J M, Lee K Y, et al. Catalytic transfer hydrogenation/hydrogenolysis of guaiacol to cyclohexane over bimetallic RuRe/C catalysts[J]. Catalysis Communications, 2016, 86: 113⁃118.
[本文引用: 7]
[62]
Wei J, José L S, Laura P P, et al. Noble metal supported on activated carbon for “hydrogen free” HDO reactions: Exploring economically advantageous routes for biomass valorisation[J]. ChemCatChem, 2019, 11(17): 4434⁃4441.
[本文引用: 2]
[63]
Yang Z, Luo B W, Shu R Y, et al. Efficient hydrodeoxygenation of phenolic compounds and raw lignin⁃oil under a temperature⁃controlled phase⁃transfer catalysis[J]. Fuel, 2021, 291: 120091⁃120100.
[本文引用: 1]
[64]
Liang X, Fan G L, Yang L, et al. Structure⁃tunable pompon⁃like RuCo catalysts: Insight into the roles of atomically dispersed Ru⁃Co sites and crystallographic structures for guaiacol hydrodeoxygenation[J]. Journal of Catalysis, 2021, 398: 76⁃88.
[本文引用: 3]
[65]
Xu Q Q, Shi Y S, Yang L, et al. The promotional effect of surface Ru decoration on the catalytic performance of Co⁃based nanocatalysts for guaiacol hydrodeoxygenation[J]. Molecular Catalysis, 2020, 497: 111224⁃111233.
[本文引用: 4]
[66]
Shu R Y, Li R X, Liu Y, et al. Enhanced adsorption properties of bimetallic RuCo catalyst for the hydrodeoxygenation of phenolic compounds and raw lignin⁃oil[J]. Chemical Engineering Science, 2020, 227: 115920⁃115955.
[本文引用: 3]
[67]
Li R X, Qiu J J, Chen H Q, et al. Hydrodeoxygenation of phenolic compounds and raw lignin⁃oil over bimetallic RuNi catalyst: An experimental and modeling study focusing on adsorption properties[J]. Fuel, 2020, 281: 118758⁃118767.
[本文引用: 3]
[68]
Long W, Liu P L, Xiong W, et al. Conversion of guaiacol as lignin model component using acid⁃treated, multi⁃walled carbon nanotubes supported Ru⁃MnO bimetallic catalysts[J]. Canadian Journal of Chemistry, 2020, 98(2): 57⁃65.
[本文引用: 4]
[69]
Lee E H, Parket R S, Kim H, et al. Hydrodeoxygenation of guaiacol over Pt loaded zeolitic materials[J]. Journal of Industrial and Engineering Chemistry, 2016, 37: 18⁃21.
[本文引用: 2]
[70]
Nie L, Peng B, Zhu X L. Vapor‐phase hydrodeoxygenation of guaiacol to aromatics over Pt/HBeta: Identification of the role of acid sites and metal sites on the reaction pathway[J]. ChemCatChem, 2018, 10(5): 1064⁃1074.
[本文引用: 2]
[71]
Silva N K G, Ferreira R A R, Ribas R M, et al. Gas⁃phase hydrodeoxygenation (HDO) of guaiacol over Pt/Al2 O3 catalyst promoted by Nb2 O5 [J]. Fuel, 2020, 287: 119509⁃119519.
[本文引用: 2]
[72]
He Z, Hu M C, Wang X Q. Highly effective hydrodeoxygenation of guaiacol on Pt/TiO2 : Promoter effects[J]. Catalysis Today, 2018, 302: 136⁃145.
[本文引用: 1]
[73]
Shafaghat H, Lee I G, Jae J, et al. Pd/C catalyzed transfer hydrogenation of pyrolysis oil using 2⁃propanol as hydrogen source[J]. Chemical Engineering Journal, 2019, 377: 119986.
[本文引用: 4]
[74]
Thompson S T, Lamb H H. Vapor⁃phase hydrodeoxygenation of guaiacol over carbon⁃supported Pd, Re and PdRe catalysts[J]. Applied Catalysis A: General, 2018, 563: 105⁃117.
[本文引用: 4]
[75]
Jeonga Y, Park C W, Park Y K, et al. Investigation of the activity and selectivity of supported rhenium catalysts for the hydrodeoxygenation of 2⁃methoxyphenol[J]. Catalysis Today, 2020, 375: 164⁃173.
[本文引用: 2]
[76]
He Y F, Bie Y W, Lehtonen J, et al. Hydrodeoxygenation of guaiacol as a model compound of lignin⁃derived pyrolysis bio⁃oil over zirconia⁃supported Rh catalyst: Process optimization and reaction kinetics[J]. Fuel, 2019, 239: 1015⁃1027.
[本文引用: 2]
[77]
Liu K R, Yan P F, Jiang H, et al. Silver initiated hydrogen spillover on anatase TiO2 creates active sites for selective hydrodeoxygenation of guaiacol[J]. Journal of Catalysis, 2019, 369: 396⁃404.
[本文引用: 2]
[78]
Mao J B, Zhou J X, Zhi X, et al. Anatase TiO2 activated by gold nanoparticles for selective hydrodeoxygenation of guaiacol to phenolics[J]. ACS Catalysis, 2017, 7(1): 695⁃705.
[本文引用: 3]
1
... 自工业革命以来,人们对石油、煤炭等化石资源的过度开采和大量使用带来了能源危机,同时也引发了严重的环境和生态问题[1 ] .在能源日趋枯竭和环境日益恶化的双重压力下,科研人员积极研发新型能源,如开发利用太阳能、风能、水能和生物质能等可再生能源[2 ] .在众多的可再生能源中,生物质能[3 ] 有其独特的优势.与太阳能、风能和水能等可再生能源不同,生物质与石油、煤炭组成相近,主要成分均由C、H元素组成,最有望成为传统化石能源的替代品.通过开发利用生物质能,可以满足人类社会对CH基能源的持久需求,有助于构建可持续发展和环境友好的能源体系. ...
1
... 自工业革命以来,人们对石油、煤炭等化石资源的过度开采和大量使用带来了能源危机,同时也引发了严重的环境和生态问题[1 ] .在能源日趋枯竭和环境日益恶化的双重压力下,科研人员积极研发新型能源,如开发利用太阳能、风能、水能和生物质能等可再生能源[2 ] .在众多的可再生能源中,生物质能[3 ] 有其独特的优势.与太阳能、风能和水能等可再生能源不同,生物质与石油、煤炭组成相近,主要成分均由C、H元素组成,最有望成为传统化石能源的替代品.通过开发利用生物质能,可以满足人类社会对CH基能源的持久需求,有助于构建可持续发展和环境友好的能源体系. ...
2
... 自工业革命以来,人们对石油、煤炭等化石资源的过度开采和大量使用带来了能源危机,同时也引发了严重的环境和生态问题[1 ] .在能源日趋枯竭和环境日益恶化的双重压力下,科研人员积极研发新型能源,如开发利用太阳能、风能、水能和生物质能等可再生能源[2 ] .在众多的可再生能源中,生物质能[3 ] 有其独特的优势.与太阳能、风能和水能等可再生能源不同,生物质与石油、煤炭组成相近,主要成分均由C、H元素组成,最有望成为传统化石能源的替代品.通过开发利用生物质能,可以满足人类社会对CH基能源的持久需求,有助于构建可持续发展和环境友好的能源体系. ...
... 生物质的主要成分包括三个部分(质量分数),即40%~50%的纤维素、20%~40%的半纤维素和20%~30%的木质素[3 ] .木质素是由苯丙烷类单元组成的复杂三维非晶态聚合物,主要由对羟基苯基、愈创木基和紫丁香基三种基本苯基结构单元以C-O和C-C等非线性随机连接组成.木质素因其独特的芳香结构,在生产液体燃料以及高附加值的六元环化学品方面具有很高的应用价值.由于木质素是三维非晶态聚合物,且其解聚产物成分复杂,现阶段有关木质素加氢精制的研究,多采用模型化合物来开展研究工作[4 ] .愈创木酚结构含有芳环和CAR -OH、CAR -OCH3 含氧官能团,这些基团是木质素结构的主要组成部分.因此,愈创木酚常被用作研究木质素催化转化的模型化合物. ...
1
... 生物质的主要成分包括三个部分(质量分数),即40%~50%的纤维素、20%~40%的半纤维素和20%~30%的木质素[3 ] .木质素是由苯丙烷类单元组成的复杂三维非晶态聚合物,主要由对羟基苯基、愈创木基和紫丁香基三种基本苯基结构单元以C-O和C-C等非线性随机连接组成.木质素因其独特的芳香结构,在生产液体燃料以及高附加值的六元环化学品方面具有很高的应用价值.由于木质素是三维非晶态聚合物,且其解聚产物成分复杂,现阶段有关木质素加氢精制的研究,多采用模型化合物来开展研究工作[4 ] .愈创木酚结构含有芳环和CAR -OH、CAR -OCH3 含氧官能团,这些基团是木质素结构的主要组成部分.因此,愈创木酚常被用作研究木质素催化转化的模型化合物. ...
4
... 图1 为木质素的模型化合物愈创木酚加氢产物分布.从图1 可以看出,以愈创木酚为原料经加氢反应能生成多种化合物.通过解离CAR -OCH3 ,能够制备苯酚,若同时解离CAR -OCH3 和将苯环加氢,就能够制备环己醇.此外,还可以制备多种化合物.在不同的催化剂和加氢脱氧反应工艺条件下,愈创木酚通过苯环氢化、烷基转移、去甲基化、去甲氧基化、甲基转移和脱水反应等方式转化为不同的产物[5 ] . ...
... 除了Ni组分,Mo、Co和Fe等过渡金属也被用来构建愈创木酚加氢反应催化剂,相关研究结果见表2 .Mo是采用较多的催化活性组分,仅次于Ni,负载在碳载体上的Mo基催化剂被广泛用于愈创木酚反应中[5 ,40 ⁃45 ] ,其产物以苯酚或苯为主.S.Ansaloni等[40 ] 在间歇式反应器中测试了Mo负载在SiO2 、Al2 O3 、NaY沸石、MgO、AC和石墨载体上的催化剂的催化活性.结果表明,在350 ℃、4.00 MPa H2 的条件下,负载在AC上的Mo基催化剂对愈创木酚脱甲氧基表现出最佳性能,愈创木酚完全转化,苯酚的收率为72.0%.AC的高表面积使MoO x x 2 C催化剂在愈创木酚的加氢脱氧反应活性[41 ⁃43 ] .Z.Cai等[45 ] 制备了AC负载的MoO2 催化剂,在300 ℃、3.00 MPa H2 的条件下研究了愈创木酚的加氢脱氧反应,得到的主要产物为酚类,且苯酚的收率达到72.0%.上述结果说明,采用以Mo为金属组分、以碳材料为载体的催化剂,产物倾向于芳香族化合物. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... 载体除了起到担载和分散活性组分的作用,还可能影响催化功能.多种类型的碳材料被广泛用作载体,例如AC、CNT和石墨烯等.相较于酸性较高的分子筛、金属氧化物载体,碳材料由于普遍缺乏酸位点,可以在一定程度上抑制焦炭形成,提高催化剂的寿命[5 ⁃6 ] .但是,由于碳基载体的酸度太低,在活化含氧基团方面显得能力较低,因此基于碳材料的催化剂通常不是依靠载体构建酸催化功能.例如,Ni5 ⁃Fe1 /CNT[32 ] 、Co⁃MoO2 /C[48 ] 、RuRe/C[61 ] 等碳基催化剂采用双金属组分,其中呈氧化态的金属助剂发挥酸催化功能.对SiO2 、Al2 O3 和分子筛等具有一定酸性的载体,酸性太弱的载体会体现酸功能不足,中度酸性载体构造的催化剂往往具有解离CAR -OCH3 的能力,可提高对苯酚的选择性[20 ⁃23 ] ,而基于强酸性载体的催化剂有可能使苯酚的羟基也脱除,得到完全脱氧产物. ...
2
... P.Sangnikul等[6 ] 采用Cu或Ce调控的NiMo/γ⁃Al2 O3 催化剂对愈创木酚进行了加氢脱氧反应研究.结果表明,通过甲基取代或烷基转移反应生成了甲氧基甲基酚和1,2⁃二甲氧基苯.T.Li等[7 ] 的研究表明,采用Ni/CK⁃800催化剂,在温度为220 ℃、氢分压为2.00 MPa、反应时间为2 h的条件下,愈创木酚完全转化,主要产物为1⁃甲基⁃1,2⁃环己二醇(收率为73.0%),得到加氢不脱氧的产物.M.H.Zhou等[8 ] 采用NiCo/γ⁃Al2 O3 催化剂对愈创木酚进行加氢部分脱氧反应,主要产物为环己醇.研究发现,愈创木酚反应过程中的中间体主要为苯酚,还有少量的1⁃甲基⁃1,2⁃环己二醇.这说明愈创木酚的加氢可能经过两个反应路径:愈创木酚先氢解甲氧基,生成苯酚,进一步转化为环己酮,最后生成环己醇;愈创木酚通过苯环直接加氢,然后异构化为1⁃甲基⁃1,2⁃环己二醇,进一步解离甲基和一个羟基,生成环己醇.I.B.Adilina等[9 ] 以NiMoPS/PILC为催化剂,在温度为350 °C、氢分压为2.00 MPa的条件下反应6 h,主要产物为苯酚,其余为邻甲酚和对甲酚.Q.H.Lai等[10 ] 在SiO2 ⁃Al2 O3 负载的金属催化剂上研究了愈创木酚的加氢脱氧途径.结果表明,愈创木酚先通过脱甲基或甲基转移生成邻苯二酚,然后邻苯二酚解离一个C-O,生成酚类化合物,进一步解离余下的C-O生成苯类化合物.S.Gutiérrez⁃Rubio等[11 ] 通过以不同的二维沸石负载磷化镍为催化剂研究了愈创木酚的加氢脱氧反应,发现了以苯甲醚为中间产物的反应路线:饱和苯甲醚的芳环生成甲氧基环己烷,进一步脱甲氧基生成环己烷;苯甲醚去甲氧基化生成苯,进而加氢生成环己烯和环己烷.因此,愈创木酚的加氢脱氧反应路径可归纳为如下四类: ...
... 载体除了起到担载和分散活性组分的作用,还可能影响催化功能.多种类型的碳材料被广泛用作载体,例如AC、CNT和石墨烯等.相较于酸性较高的分子筛、金属氧化物载体,碳材料由于普遍缺乏酸位点,可以在一定程度上抑制焦炭形成,提高催化剂的寿命[5 ⁃6 ] .但是,由于碳基载体的酸度太低,在活化含氧基团方面显得能力较低,因此基于碳材料的催化剂通常不是依靠载体构建酸催化功能.例如,Ni5 ⁃Fe1 /CNT[32 ] 、Co⁃MoO2 /C[48 ] 、RuRe/C[61 ] 等碳基催化剂采用双金属组分,其中呈氧化态的金属助剂发挥酸催化功能.对SiO2 、Al2 O3 和分子筛等具有一定酸性的载体,酸性太弱的载体会体现酸功能不足,中度酸性载体构造的催化剂往往具有解离CAR -OCH3 的能力,可提高对苯酚的选择性[20 ⁃23 ] ,而基于强酸性载体的催化剂有可能使苯酚的羟基也脱除,得到完全脱氧产物. ...
5
... P.Sangnikul等[6 ] 采用Cu或Ce调控的NiMo/γ⁃Al2 O3 催化剂对愈创木酚进行了加氢脱氧反应研究.结果表明,通过甲基取代或烷基转移反应生成了甲氧基甲基酚和1,2⁃二甲氧基苯.T.Li等[7 ] 的研究表明,采用Ni/CK⁃800催化剂,在温度为220 ℃、氢分压为2.00 MPa、反应时间为2 h的条件下,愈创木酚完全转化,主要产物为1⁃甲基⁃1,2⁃环己二醇(收率为73.0%),得到加氢不脱氧的产物.M.H.Zhou等[8 ] 采用NiCo/γ⁃Al2 O3 催化剂对愈创木酚进行加氢部分脱氧反应,主要产物为环己醇.研究发现,愈创木酚反应过程中的中间体主要为苯酚,还有少量的1⁃甲基⁃1,2⁃环己二醇.这说明愈创木酚的加氢可能经过两个反应路径:愈创木酚先氢解甲氧基,生成苯酚,进一步转化为环己酮,最后生成环己醇;愈创木酚通过苯环直接加氢,然后异构化为1⁃甲基⁃1,2⁃环己二醇,进一步解离甲基和一个羟基,生成环己醇.I.B.Adilina等[9 ] 以NiMoPS/PILC为催化剂,在温度为350 °C、氢分压为2.00 MPa的条件下反应6 h,主要产物为苯酚,其余为邻甲酚和对甲酚.Q.H.Lai等[10 ] 在SiO2 ⁃Al2 O3 负载的金属催化剂上研究了愈创木酚的加氢脱氧途径.结果表明,愈创木酚先通过脱甲基或甲基转移生成邻苯二酚,然后邻苯二酚解离一个C-O,生成酚类化合物,进一步解离余下的C-O生成苯类化合物.S.Gutiérrez⁃Rubio等[11 ] 通过以不同的二维沸石负载磷化镍为催化剂研究了愈创木酚的加氢脱氧反应,发现了以苯甲醚为中间产物的反应路线:饱和苯甲醚的芳环生成甲氧基环己烷,进一步脱甲氧基生成环己烷;苯甲醚去甲氧基化生成苯,进而加氢生成环己烯和环己烷.因此,愈创木酚的加氢脱氧反应路径可归纳为如下四类: ...
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... [7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ]和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 载体是催化剂的重要组成部分,不仅起到分散活性组分的作用,还会影响催化剂的反应活性、催化反应的路径以及催化剂的寿命.关于愈创木酚加氢反应的催化剂,研究者尝试了多种载体材料,例如以活性炭(AC)、CNT、碳纳米纤维(CNF)和石墨烯等为代表的碳材料以及SiO2 、ZrO2 、CeO2 、TiO2 和γ⁃Al2 O3 等.研究表明,以碳材料为载体的Ni基催化剂对酚类加氢脱氧具有很高的活性,可以使愈创木酚更趋向于苯环加氢反应,产物多为环己醇、环己烷、2⁃甲氧基环己醇、1⁃甲基⁃1,2⁃环己二醇等产品.T.Li等[7 ] 考察了碳化温度和促进剂对催化剂加氢脱氧性能的影响.结果表明,800 ℃碳化的Ni/CK⁃800催化剂在NaBH4 溶液中还原后,在180 ℃、2.00 MPa H2 、2 h的条件下,对愈创木酚转化表现出优异的活性,1⁃甲基⁃1,2⁃环己二醇的收率为73.0%.研究发现,通过增加催化剂酸度和降低催化剂表面C-O基团的含量,有利于提高愈创木酚的转化率和对1⁃甲基⁃1,2⁃环己二醇的选择性.分子筛和氧化物类载体会影响催化剂的酸性,对催化性能起重要作用.M.H.Zhou等[8 ] 在200 ℃、5.00 MPa H2 、8 h的反应条件下,考察了γ⁃Al2 O3 和ZSM⁃5负载的NiCo基催化剂对愈创木酚的加氢脱氧反应.结果表明,NiCo/γ⁃Al2 O3 催化剂比NiCo/ZSM⁃5催化剂酸性弱,而当使用ZSM⁃5作为载体时,由于其酸性较强,导致环己烷的选择性增加.由此可以看出,增加酸性有利于环己醇脱水为环己烷,从而改变产物的选择性. ...
10
... P.Sangnikul等[6 ] 采用Cu或Ce调控的NiMo/γ⁃Al2 O3 催化剂对愈创木酚进行了加氢脱氧反应研究.结果表明,通过甲基取代或烷基转移反应生成了甲氧基甲基酚和1,2⁃二甲氧基苯.T.Li等[7 ] 的研究表明,采用Ni/CK⁃800催化剂,在温度为220 ℃、氢分压为2.00 MPa、反应时间为2 h的条件下,愈创木酚完全转化,主要产物为1⁃甲基⁃1,2⁃环己二醇(收率为73.0%),得到加氢不脱氧的产物.M.H.Zhou等[8 ] 采用NiCo/γ⁃Al2 O3 催化剂对愈创木酚进行加氢部分脱氧反应,主要产物为环己醇.研究发现,愈创木酚反应过程中的中间体主要为苯酚,还有少量的1⁃甲基⁃1,2⁃环己二醇.这说明愈创木酚的加氢可能经过两个反应路径:愈创木酚先氢解甲氧基,生成苯酚,进一步转化为环己酮,最后生成环己醇;愈创木酚通过苯环直接加氢,然后异构化为1⁃甲基⁃1,2⁃环己二醇,进一步解离甲基和一个羟基,生成环己醇.I.B.Adilina等[9 ] 以NiMoPS/PILC为催化剂,在温度为350 °C、氢分压为2.00 MPa的条件下反应6 h,主要产物为苯酚,其余为邻甲酚和对甲酚.Q.H.Lai等[10 ] 在SiO2 ⁃Al2 O3 负载的金属催化剂上研究了愈创木酚的加氢脱氧途径.结果表明,愈创木酚先通过脱甲基或甲基转移生成邻苯二酚,然后邻苯二酚解离一个C-O,生成酚类化合物,进一步解离余下的C-O生成苯类化合物.S.Gutiérrez⁃Rubio等[11 ] 通过以不同的二维沸石负载磷化镍为催化剂研究了愈创木酚的加氢脱氧反应,发现了以苯甲醚为中间产物的反应路线:饱和苯甲醚的芳环生成甲氧基环己烷,进一步脱甲氧基生成环己烷;苯甲醚去甲氧基化生成苯,进而加氢生成环己烯和环己烷.因此,愈创木酚的加氢脱氧反应路径可归纳为如下四类: ...
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ]和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
... [8 ]通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
... 载体是催化剂的重要组成部分,不仅起到分散活性组分的作用,还会影响催化剂的反应活性、催化反应的路径以及催化剂的寿命.关于愈创木酚加氢反应的催化剂,研究者尝试了多种载体材料,例如以活性炭(AC)、CNT、碳纳米纤维(CNF)和石墨烯等为代表的碳材料以及SiO2 、ZrO2 、CeO2 、TiO2 和γ⁃Al2 O3 等.研究表明,以碳材料为载体的Ni基催化剂对酚类加氢脱氧具有很高的活性,可以使愈创木酚更趋向于苯环加氢反应,产物多为环己醇、环己烷、2⁃甲氧基环己醇、1⁃甲基⁃1,2⁃环己二醇等产品.T.Li等[7 ] 考察了碳化温度和促进剂对催化剂加氢脱氧性能的影响.结果表明,800 ℃碳化的Ni/CK⁃800催化剂在NaBH4 溶液中还原后,在180 ℃、2.00 MPa H2 、2 h的条件下,对愈创木酚转化表现出优异的活性,1⁃甲基⁃1,2⁃环己二醇的收率为73.0%.研究发现,通过增加催化剂酸度和降低催化剂表面C-O基团的含量,有利于提高愈创木酚的转化率和对1⁃甲基⁃1,2⁃环己二醇的选择性.分子筛和氧化物类载体会影响催化剂的酸性,对催化性能起重要作用.M.H.Zhou等[8 ] 在200 ℃、5.00 MPa H2 、8 h的反应条件下,考察了γ⁃Al2 O3 和ZSM⁃5负载的NiCo基催化剂对愈创木酚的加氢脱氧反应.结果表明,NiCo/γ⁃Al2 O3 催化剂比NiCo/ZSM⁃5催化剂酸性弱,而当使用ZSM⁃5作为载体时,由于其酸性较强,导致环己烷的选择性增加.由此可以看出,增加酸性有利于环己醇脱水为环己烷,从而改变产物的选择性. ...
... 反应温度不仅会影响催化剂的愈创木酚加氢脱氧反应活性,而且与产物选择性也有很大的关系.一般而言,对于愈创木酚的加氢脱氧反应,随着反应温度升高,反应活性增加,随之而来的是沿着不同反应路径加氢程度逐渐增加,最后归一到环己烷,再进一步反应开环得到烷烃产物.M.H.Zhou等[8 ] 在不同反应温度下探究了愈创木酚的加氢脱氧反应.结果表明,当反应温度从160 ℃增加到200 ℃时,主产物环己醇的选择性逐渐升高,反应温度为200 ℃时达到最大值;随着反应温度进一步增加至220 ℃,环己醇的选择性下降,而1⁃甲基⁃1,2⁃环己二醇(反式和顺式)的选择性有所增加.这说明反应温度影响反应路径,改变产物分布.对该反应体系,调变反应温度时需要同时考虑热力学和动力学因素,这样才有可能获得较好的产物选择性. ...
... 在愈创木酚的加氢脱氧反应中,大部分研究人员选择H2 作为H供体.H2 是反应物,氢分压的增加提高H2 在溶液中的溶解度,进而增加催化剂活性位上活性氢的浓度.研究表明[8 ,65 ] ,氢分压不仅影响加氢程度,随着反应的进行,更倾向于沿着愈创木酚―苯酚―环己醇―环己烷的方向递进,而且还会改变反应路径.M.H.Zhou等[8 ] 基于NiCo/γ⁃Al2 O3 催化剂研究了初始氢分压的影响.结果表明,随着初始氢分压增加,环己醇选择性逐渐降低,同时1⁃甲基⁃1,2⁃环己二醇(反式和顺式)的总选择性增加.苯环加氢反应需要较多的活性氢,所以当氢分压增加时,愈创木酚更容易通过加氢苯环生成2⁃甲氧基环己醇,进而异构化生成1⁃甲基⁃1,2⁃环己二醇(反式和顺式).该反应路线消耗较多的愈创木酚,使愈创木酚去甲氧基生成苯酚,苯酚再进一步加氢生成环己醇的反应被削弱.Q.Q.Xu等[65 ] 利用Ru⁃Co/C催化剂探究氢分压(0.50~2.00 MPa)对愈创木酚加氢脱氧反应(200 ℃、1.5 h)的影响.结果表明,在0.5 MPa的氢分压下,生成了大量的苯酚(收率为29.4%).因此,较低的氢分压能够降低苯环饱和反应的可能性,提高苯酚选择性. ...
... [8 ]基于NiCo/γ⁃Al2 O3 催化剂研究了初始氢分压的影响.结果表明,随着初始氢分压增加,环己醇选择性逐渐降低,同时1⁃甲基⁃1,2⁃环己二醇(反式和顺式)的总选择性增加.苯环加氢反应需要较多的活性氢,所以当氢分压增加时,愈创木酚更容易通过加氢苯环生成2⁃甲氧基环己醇,进而异构化生成1⁃甲基⁃1,2⁃环己二醇(反式和顺式).该反应路线消耗较多的愈创木酚,使愈创木酚去甲氧基生成苯酚,苯酚再进一步加氢生成环己醇的反应被削弱.Q.Q.Xu等[65 ] 利用Ru⁃Co/C催化剂探究氢分压(0.50~2.00 MPa)对愈创木酚加氢脱氧反应(200 ℃、1.5 h)的影响.结果表明,在0.5 MPa的氢分压下,生成了大量的苯酚(收率为29.4%).因此,较低的氢分压能够降低苯环饱和反应的可能性,提高苯酚选择性. ...
6
... P.Sangnikul等[6 ] 采用Cu或Ce调控的NiMo/γ⁃Al2 O3 催化剂对愈创木酚进行了加氢脱氧反应研究.结果表明,通过甲基取代或烷基转移反应生成了甲氧基甲基酚和1,2⁃二甲氧基苯.T.Li等[7 ] 的研究表明,采用Ni/CK⁃800催化剂,在温度为220 ℃、氢分压为2.00 MPa、反应时间为2 h的条件下,愈创木酚完全转化,主要产物为1⁃甲基⁃1,2⁃环己二醇(收率为73.0%),得到加氢不脱氧的产物.M.H.Zhou等[8 ] 采用NiCo/γ⁃Al2 O3 催化剂对愈创木酚进行加氢部分脱氧反应,主要产物为环己醇.研究发现,愈创木酚反应过程中的中间体主要为苯酚,还有少量的1⁃甲基⁃1,2⁃环己二醇.这说明愈创木酚的加氢可能经过两个反应路径:愈创木酚先氢解甲氧基,生成苯酚,进一步转化为环己酮,最后生成环己醇;愈创木酚通过苯环直接加氢,然后异构化为1⁃甲基⁃1,2⁃环己二醇,进一步解离甲基和一个羟基,生成环己醇.I.B.Adilina等[9 ] 以NiMoPS/PILC为催化剂,在温度为350 °C、氢分压为2.00 MPa的条件下反应6 h,主要产物为苯酚,其余为邻甲酚和对甲酚.Q.H.Lai等[10 ] 在SiO2 ⁃Al2 O3 负载的金属催化剂上研究了愈创木酚的加氢脱氧途径.结果表明,愈创木酚先通过脱甲基或甲基转移生成邻苯二酚,然后邻苯二酚解离一个C-O,生成酚类化合物,进一步解离余下的C-O生成苯类化合物.S.Gutiérrez⁃Rubio等[11 ] 通过以不同的二维沸石负载磷化镍为催化剂研究了愈创木酚的加氢脱氧反应,发现了以苯甲醚为中间产物的反应路线:饱和苯甲醚的芳环生成甲氧基环己烷,进一步脱甲氧基生成环己烷;苯甲醚去甲氧基化生成苯,进而加氢生成环己烯和环己烷.因此,愈创木酚的加氢脱氧反应路径可归纳为如下四类: ...
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... [9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ]和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
... 对于串联反应,通过控制反应时间,从动力学上可获得某一中间步骤的产物.在愈创木酚的加氢脱氧反应中,一条典型的反应路径是愈创木酚―苯酚―环己醇―环己烷,随着反应时间的增加,总是由一个产物推进到另一个产物,适当控制反应时间能够提高苯酚、环己醇等中间产物的收率[9 ,25 ,34 ,40 ,45 ] . ...
4
... P.Sangnikul等[6 ] 采用Cu或Ce调控的NiMo/γ⁃Al2 O3 催化剂对愈创木酚进行了加氢脱氧反应研究.结果表明,通过甲基取代或烷基转移反应生成了甲氧基甲基酚和1,2⁃二甲氧基苯.T.Li等[7 ] 的研究表明,采用Ni/CK⁃800催化剂,在温度为220 ℃、氢分压为2.00 MPa、反应时间为2 h的条件下,愈创木酚完全转化,主要产物为1⁃甲基⁃1,2⁃环己二醇(收率为73.0%),得到加氢不脱氧的产物.M.H.Zhou等[8 ] 采用NiCo/γ⁃Al2 O3 催化剂对愈创木酚进行加氢部分脱氧反应,主要产物为环己醇.研究发现,愈创木酚反应过程中的中间体主要为苯酚,还有少量的1⁃甲基⁃1,2⁃环己二醇.这说明愈创木酚的加氢可能经过两个反应路径:愈创木酚先氢解甲氧基,生成苯酚,进一步转化为环己酮,最后生成环己醇;愈创木酚通过苯环直接加氢,然后异构化为1⁃甲基⁃1,2⁃环己二醇,进一步解离甲基和一个羟基,生成环己醇.I.B.Adilina等[9 ] 以NiMoPS/PILC为催化剂,在温度为350 °C、氢分压为2.00 MPa的条件下反应6 h,主要产物为苯酚,其余为邻甲酚和对甲酚.Q.H.Lai等[10 ] 在SiO2 ⁃Al2 O3 负载的金属催化剂上研究了愈创木酚的加氢脱氧途径.结果表明,愈创木酚先通过脱甲基或甲基转移生成邻苯二酚,然后邻苯二酚解离一个C-O,生成酚类化合物,进一步解离余下的C-O生成苯类化合物.S.Gutiérrez⁃Rubio等[11 ] 通过以不同的二维沸石负载磷化镍为催化剂研究了愈创木酚的加氢脱氧反应,发现了以苯甲醚为中间产物的反应路线:饱和苯甲醚的芳环生成甲氧基环己烷,进一步脱甲氧基生成环己烷;苯甲醚去甲氧基化生成苯,进而加氢生成环己烯和环己烷.因此,愈创木酚的加氢脱氧反应路径可归纳为如下四类: ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
... 单金属Pd催化剂[73 ] 及PdNi[10 ] 、PdFe[53 ⁃54 ] 、PdRe[74 ] 双金属催化剂也被用于愈创木酚的加氢脱氧反应.基于Pd催化剂的产物种类也很多,有的以芳香族化合物为主[10 ,53 ,74 ] ,有的是芳环加氢饱和的产物[73 ] .H.Shafaghat等[73 ] 以Pd/C为催化剂,探究了醇类(甲醇、乙醇、1⁃丙醇、1⁃丁醇、异丙醇和2⁃丁醇等)替代H2 的可行性.结果表明,只有仲醇可以作为Pd/C上愈创木酚转化的H供体.同时,当使用异丙醇作为H供体时,生成较多的1⁃甲丙基环己烷(收率为49.3%).S.T.Thompson等[74 ] 采用Re对Pd/C催化剂进行修饰,结果表明,在400 ℃原位还原后,PdRe/C可获得完全脱氧产物(苯和环己烷),其中苯的收率为35.0%.PdRe双金属催化剂结合了Pd/C的脱甲氧基和加氢功能与Re/C的对苯酚的脱氧功能. ...
... [10 ,53 ,74 ],有的是芳环加氢饱和的产物[73 ] .H.Shafaghat等[73 ] 以Pd/C为催化剂,探究了醇类(甲醇、乙醇、1⁃丙醇、1⁃丁醇、异丙醇和2⁃丁醇等)替代H2 的可行性.结果表明,只有仲醇可以作为Pd/C上愈创木酚转化的H供体.同时,当使用异丙醇作为H供体时,生成较多的1⁃甲丙基环己烷(收率为49.3%).S.T.Thompson等[74 ] 采用Re对Pd/C催化剂进行修饰,结果表明,在400 ℃原位还原后,PdRe/C可获得完全脱氧产物(苯和环己烷),其中苯的收率为35.0%.PdRe双金属催化剂结合了Pd/C的脱甲氧基和加氢功能与Re/C的对苯酚的脱氧功能. ...
5
... P.Sangnikul等[6 ] 采用Cu或Ce调控的NiMo/γ⁃Al2 O3 催化剂对愈创木酚进行了加氢脱氧反应研究.结果表明,通过甲基取代或烷基转移反应生成了甲氧基甲基酚和1,2⁃二甲氧基苯.T.Li等[7 ] 的研究表明,采用Ni/CK⁃800催化剂,在温度为220 ℃、氢分压为2.00 MPa、反应时间为2 h的条件下,愈创木酚完全转化,主要产物为1⁃甲基⁃1,2⁃环己二醇(收率为73.0%),得到加氢不脱氧的产物.M.H.Zhou等[8 ] 采用NiCo/γ⁃Al2 O3 催化剂对愈创木酚进行加氢部分脱氧反应,主要产物为环己醇.研究发现,愈创木酚反应过程中的中间体主要为苯酚,还有少量的1⁃甲基⁃1,2⁃环己二醇.这说明愈创木酚的加氢可能经过两个反应路径:愈创木酚先氢解甲氧基,生成苯酚,进一步转化为环己酮,最后生成环己醇;愈创木酚通过苯环直接加氢,然后异构化为1⁃甲基⁃1,2⁃环己二醇,进一步解离甲基和一个羟基,生成环己醇.I.B.Adilina等[9 ] 以NiMoPS/PILC为催化剂,在温度为350 °C、氢分压为2.00 MPa的条件下反应6 h,主要产物为苯酚,其余为邻甲酚和对甲酚.Q.H.Lai等[10 ] 在SiO2 ⁃Al2 O3 负载的金属催化剂上研究了愈创木酚的加氢脱氧途径.结果表明,愈创木酚先通过脱甲基或甲基转移生成邻苯二酚,然后邻苯二酚解离一个C-O,生成酚类化合物,进一步解离余下的C-O生成苯类化合物.S.Gutiérrez⁃Rubio等[11 ] 通过以不同的二维沸石负载磷化镍为催化剂研究了愈创木酚的加氢脱氧反应,发现了以苯甲醚为中间产物的反应路线:饱和苯甲醚的芳环生成甲氧基环己烷,进一步脱甲氧基生成环己烷;苯甲醚去甲氧基化生成苯,进而加氢生成环己烯和环己烷.因此,愈创木酚的加氢脱氧反应路径可归纳为如下四类: ...
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... [11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ]为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
5
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ]和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ]和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... [
12 ]
4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
1
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
3
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,14 ⁃15 ,18 ,23 ⁃25 ,37 ]和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
2
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
4
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ]为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... [
16 ]
9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
3
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... [17 ,25 ,32 ,35 ]的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
2
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
3
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,19 ,26 ,29 ⁃30 ,32 ,34 ]和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
5
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... [20 ,28 ,36 ]为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
... 载体除了起到担载和分散活性组分的作用,还可能影响催化功能.多种类型的碳材料被广泛用作载体,例如AC、CNT和石墨烯等.相较于酸性较高的分子筛、金属氧化物载体,碳材料由于普遍缺乏酸位点,可以在一定程度上抑制焦炭形成,提高催化剂的寿命[5 ⁃6 ] .但是,由于碳基载体的酸度太低,在活化含氧基团方面显得能力较低,因此基于碳材料的催化剂通常不是依靠载体构建酸催化功能.例如,Ni5 ⁃Fe1 /CNT[32 ] 、Co⁃MoO2 /C[48 ] 、RuRe/C[61 ] 等碳基催化剂采用双金属组分,其中呈氧化态的金属助剂发挥酸催化功能.对SiO2 、Al2 O3 和分子筛等具有一定酸性的载体,酸性太弱的载体会体现酸功能不足,中度酸性载体构造的催化剂往往具有解离CAR -OCH3 的能力,可提高对苯酚的选择性[20 ⁃23 ] ,而基于强酸性载体的催化剂有可能使苯酚的羟基也脱除,得到完全脱氧产物. ...
3
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... R.N.Olcese等[52 ] 探索了Fe/SiO2 对愈创木酚的加氢脱氧反应,发现负载15.0%的Fe催化剂在愈创木酚的加氢脱氧中对苯环的加氢反应具有惰性,可以将反应控制在产物为芳香族化合物,从而防止愈创木酚过度加氢.基于Fe/CeO2 [51 ] 和Fe/AC[21 ] 的反应,产物主要也是芳香族化合物,可见基于Fe催化剂可以选择性催化芳基醚键的断裂,有利于生成芳香类产物.然而,Fe基催化剂的催化活性较低,需要在300~400 ℃的高温条件下才能转化.为了克服这一缺陷,通常引入另一种金属,以显著提高催化性能[32 ,53 ⁃54 ] .例如,N.T.T.Tran等[53 ] 以Pd⁃Fe/Al⁃MCM⁃41为催化剂将愈创木酚转化为苯酚,其收率约为50.0%. ...
4
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,22 ,30 ⁃33 ,35 ,39 ]为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
5
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,23 ⁃25 ,37 ]和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
... 载体除了起到担载和分散活性组分的作用,还可能影响催化功能.多种类型的碳材料被广泛用作载体,例如AC、CNT和石墨烯等.相较于酸性较高的分子筛、金属氧化物载体,碳材料由于普遍缺乏酸位点,可以在一定程度上抑制焦炭形成,提高催化剂的寿命[5 ⁃6 ] .但是,由于碳基载体的酸度太低,在活化含氧基团方面显得能力较低,因此基于碳材料的催化剂通常不是依靠载体构建酸催化功能.例如,Ni5 ⁃Fe1 /CNT[32 ] 、Co⁃MoO2 /C[48 ] 、RuRe/C[61 ] 等碳基催化剂采用双金属组分,其中呈氧化态的金属助剂发挥酸催化功能.对SiO2 、Al2 O3 和分子筛等具有一定酸性的载体,酸性太弱的载体会体现酸功能不足,中度酸性载体构造的催化剂往往具有解离CAR -OCH3 的能力,可提高对苯酚的选择性[20 ⁃23 ] ,而基于强酸性载体的催化剂有可能使苯酚的羟基也脱除,得到完全脱氧产物. ...
6
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
... [24 ]通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
7
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ⁃25 ,37 ]和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,25 ,32 ,35 ]的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
... [25 ]采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
... 对于串联反应,通过控制反应时间,从动力学上可获得某一中间步骤的产物.在愈创木酚的加氢脱氧反应中,一条典型的反应路径是愈创木酚―苯酚―环己醇―环己烷,随着反应时间的增加,总是由一个产物推进到另一个产物,适当控制反应时间能够提高苯酚、环己醇等中间产物的收率[9 ,25 ,34 ,40 ,45 ] . ...
5
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,26 ]的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,26 ,29 ⁃30 ,32 ,34 ]和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
2
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
3
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,28 ,36 ]为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
2
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
5
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ⁃30 ,32 ,34 ]和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,30 ⁃33 ,35 ,39 ]为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
3
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
9
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,32 ,34 ]和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,32 ,35 ]的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... [
32 ]
29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
... [32 ]以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
... R.N.Olcese等[52 ] 探索了Fe/SiO2 对愈创木酚的加氢脱氧反应,发现负载15.0%的Fe催化剂在愈创木酚的加氢脱氧中对苯环的加氢反应具有惰性,可以将反应控制在产物为芳香族化合物,从而防止愈创木酚过度加氢.基于Fe/CeO2 [51 ] 和Fe/AC[21 ] 的反应,产物主要也是芳香族化合物,可见基于Fe催化剂可以选择性催化芳基醚键的断裂,有利于生成芳香类产物.然而,Fe基催化剂的催化活性较低,需要在300~400 ℃的高温条件下才能转化.为了克服这一缺陷,通常引入另一种金属,以显著提高催化性能[32 ,53 ⁃54 ] .例如,N.T.T.Tran等[53 ] 以Pd⁃Fe/Al⁃MCM⁃41为催化剂将愈创木酚转化为苯酚,其收率约为50.0%. ...
... 载体除了起到担载和分散活性组分的作用,还可能影响催化功能.多种类型的碳材料被广泛用作载体,例如AC、CNT和石墨烯等.相较于酸性较高的分子筛、金属氧化物载体,碳材料由于普遍缺乏酸位点,可以在一定程度上抑制焦炭形成,提高催化剂的寿命[5 ⁃6 ] .但是,由于碳基载体的酸度太低,在活化含氧基团方面显得能力较低,因此基于碳材料的催化剂通常不是依靠载体构建酸催化功能.例如,Ni5 ⁃Fe1 /CNT[32 ] 、Co⁃MoO2 /C[48 ] 、RuRe/C[61 ] 等碳基催化剂采用双金属组分,其中呈氧化态的金属助剂发挥酸催化功能.对SiO2 、Al2 O3 和分子筛等具有一定酸性的载体,酸性太弱的载体会体现酸功能不足,中度酸性载体构造的催化剂往往具有解离CAR -OCH3 的能力,可提高对苯酚的选择性[20 ⁃23 ] ,而基于强酸性载体的催化剂有可能使苯酚的羟基也脱除,得到完全脱氧产物. ...
5
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... [33 ]以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ⁃33 ,35 ,39 ]为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 一些Ni基催化剂,通过P[11 ,20 ⁃22 ] 、B[23 ] 非金属组分和Co[8 ,24 ⁃25 ] 、Mo[9 ,26 ⁃30 ] 、Fe[31 ⁃32 ] 、Cu[33 ] 等金属组分进行调控.例如,C.Z.Chen等[25 ] 采用双金属NiCo/CNT催化剂催化了愈创木酚的加氢脱氧反应.结果表明,基于Ni/CNT催化剂的反应获得愈创木酚的转化率很低(约40.0%),加氢脱氧产物以苯酚和环己醇为主;在催化剂中引入Co组分后,NiCo/CNT双金属催化剂使愈创木酚的转化率提高到100.0%,而且环己醇选择性非常高,在液体产物中只检测到非常少量的环己烷.M.H.Zhou等[8 ] 通过引入金属Co调节了催化剂的酸度.结果表明,与Ni/γ⁃Al2 O3 催化剂相比,在相同的条件下NiCo/γ⁃Al2 O3 催化剂活性更高,环己醇为主产物.这是因为Co的引入提高了催化剂的酸度、还原性和金属颗粒分散度,从而提高了催化活性.E.Blanco等[24 ] 通过浸渍法制备了负载于高表面积石墨上的单金属Ni、Co及其双金属催化剂,并将其用于愈创木酚的加氢脱氧反应.结果表明,在氢分压为5.00 MPa和反应温度为300 ℃的条件下,Ni基催化剂的加氢活性很高,倾向于将苯酚加氢生成环己醇,而催化剂中引入Co后加氢活性在一定程度上受到抑制,Ni和Co之间的协同作用抑制了环己醇的形成,从而提高了苯酚的选择性.H.H.Fang等[32 ] 以Fe为促进剂,制备了负载在碳纳米管(CNT)上的Ni基催化剂.结果表明,通过调节Ni和Fe的比例能有效控制产物分布. ...
4
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,34 ]和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
... 对于串联反应,通过控制反应时间,从动力学上可获得某一中间步骤的产物.在愈创木酚的加氢脱氧反应中,一条典型的反应路径是愈创木酚―苯酚―环己醇―环己烷,随着反应时间的增加,总是由一个产物推进到另一个产物,适当控制反应时间能够提高苯酚、环己醇等中间产物的收率[9 ,25 ,34 ,40 ,45 ] . ...
4
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,35 ,39 ]为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,35 ]的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
3
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,36 ]为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
3
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,37 ]和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
2
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
3
... 过渡金属Ni对加氢反应具有较高的催化活性,因此Ni基催化剂被广泛用于愈创木酚的加氢脱氧反应.表1 为用于愈创木酚加氢脱氧反应的Ni基催化剂.由表1 可知,单组分Ni基催化剂[7 ,12 ⁃19 ] ,P[11 ,20 ⁃22 ] 和B[23 ] 掺杂的Ni基催化剂,以及NiCo[8 ,24 ⁃25 ] 、NiMo[9 ,26 ⁃30 ] 、NiFe[31 ⁃32 ] 、NiCu[33 ] 、NiGa[34 ] 、NiRe[35 ⁃36 ] 、NiZr[37 ] 、NiCe[38 ] 、NiW[39 ] 等双组分催化剂在愈创木酚的加氢脱氧反应中均体现催化活性.大部分催化剂在投入反应体系前,需要经过H2 还原预处理,其目的是产生有活化H2 功能的金属态Ni组分.这些催化剂的反应条件差别较大,温度最低为150 ℃,最高达到410 ℃;氢分压也波动比较大,有的在1.00 MPa以下,有的高达10.00 MPa;依据催化剂活性,反应时间有的只需要1 h,有的达到10 h.在所采用的反应条件下,一多半的催化剂能使愈创木酚的转化率达到90.0%以上,说明大部分催化剂的活性较高.从反应条件来看,一些反应存在反应温度过高[27 ⁃28 ] 、氢分压过大[33 ] 以及反应时间过长[14 ,26 ] 的问题.愈创木酚在Ni基催化剂上的加氢脱氧产物比较复杂,主要有两类:以苯酚[9 ,12 ,16 ,19 ,26 ,29 ⁃30 ,32 ,34 ] 和苯[20 ,28 ,36 ] 为主的芳香族化合物、以环己醇[7 ⁃8 ,12 ,14 ⁃15 ,18 ,23 ⁃25 ,37 ] 和环己烷[11 ,16 ⁃17 ,22 ,30 ⁃33 ,35 ,39 ] 为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... ,39 ]为代表的苯环加氢产品.总的来说,产物组成随催化剂类型和反应条件而变化.然而,在表1 所列出的反应结果中,只有约1/10反应[17 ,25 ,32 ,35 ] 的主要产物收率不低于90.0%,说明催化选择性是本反应的技术难题.若选择性低,则降低主要产物收率,也增加产物分离提纯成本.因此,选择性是衡量催化剂性能的重要指标. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ni/CK⁃800 200 2 2.00 十二烷 100.0 1⁃甲基⁃1,2⁃己二醇(73.0) [7 ] 2 Ni/TiO2 ⁃R H2 /Ar/400 ℃ 300 2 4.00 正十烷 89.1 2⁃甲氧基环己醇(51.6) [12 ] 3 Ni/TiO2 ⁃A H2 /Ar/400 ℃ 300 2 4.00 正十烷 31.6 苯酚(25.6) [12 ] 4 Ni/PANI⁃AC H2 /N2 /350 ℃ 250 4 5.00 H2 O 18.0 邻苯二酚(2.6) [13 ] 5 NP⁃NiMnO2 150 12 0.50 H2 O 100.0 环己醇(75.0) [14 ] 6 Ni/ZrO2 ⁃CeO2 H2 /450 ℃ 220 3 2.00 100.0 环己醇(85.0) [15 ] 7 Ni/Al2 O3 300 4 4.00 正十烷 100.0 环己烷(80.0) [16 ] 8 Ni/TiO2 ⁃A H2 /400 ℃ 300 4 4.00 正十烷 85.0 苯酚(59.5) [16 ] 9 Ni/SiO2 ⁃TiO2 H2 /450 ℃ 220 3 2.00 正十烷 100.0 环己烷(92.0) [17 ] 10 NP⁃Ni Alloy 180 4 2.00 H2 O 99.7 环己醇(89.8) [18 ] 11 Ni/RH⁃API N2 /H2 /500 ℃ 350 1 0.10 100.0 苯酚(61.5) [19 ] 12 Ni2 P/ZSM⁃5 H2 /650 ℃ 220 2 4.00 十氢萘 78.0 环己烷(60.0) [11 ] 13a Ni2 P/SiO2 H2 /450 ℃ 350 0.10 72.0 苯(52.0) [20 ] 14a Ni2 P/γ⁃Al2 O3 H2 /Ar 300 0.10 96.9 1,2⁃二甲氧基苯(57.1) [21 ] 15 Ni2 P/HZSM⁃5 H2 /600 ℃ 300 5 3.00 十二烷 98.0 环己烷(78.8) [22 ] 16 NiB/SiO2 ⁃Al2 O3 H2 /460 ℃ 260 3 5.00 十氢萘+H2 O 98.7 环己醇(55.0) [23 ] 17 NiCo/G H2 /300 ℃ 300 4 5.00 十二烷 75.0 环己醇(37.5) [24 ] 18 NiCo/γ⁃Al2 O3 H2 200 8 5.00 H2 O 96.1 环己醇(68.1) [8 ] 19b NiCo/CNT H2 /500 ℃ 220 2 异丙醇 100.0 环己醇(94.0) [25 ] 20 NiMo/ZrO2 ⁃Al2 O3 H2 /300 ℃ 330 10 3.00 四氢萘 100.0 苯酚(45.3) [26 ] 21a NiMoW⁃磺化 H2 /H2 S/400 ℃ 400 2.80 99.6 邻苯二酚(50.0) [27 ] 22 NiMoPS/PILC H2 /500 ℃ 350 6 2.00 100.0 苯酚(77.0) [9 ] 23a NiMo/SiO2 H2 /450 ℃ 410 0.10 99.0 苯(64.0) [28 ] 24a NiMo/SBA⁃15 H2 /500 ℃ 300 85.0 苯酚(28.9) [29 ] 25 NiMoS2 /CMK⁃3 H2 /450 ℃ 300 6 5.00 十二烷 100.0 苯酚(31.5)、环己烷(35.7) [30 ] 26 FeNi/ZrO2 H2 /500 ℃ 300 8 4.00 辛烷 100.0 环己烷(89.4) [31 ] 27a Ni5 Fe1 /CNT H2 /N2 /400 ℃ 300 3.00 100.0 环己烷(99.8) [32 ] 28a Ni1 Fe5 /CNT H2 /N2 /400 ℃ 300 3.00 50.0 苯酚(42.0) [32 ] 29 NiCu/Ti⁃MCM⁃41 H2 /He/550 ℃ 260 6 10.00 庚烷 74.2 环己烷(36.2) [33 ] 30a NiGa/SiO2 300 0.10 98.4 苯酚(71.0) [34 ] 31 Ni⁃Re/SiO2 H2 /N2 /350 ℃ 250 5 3.00 十二烷 100.0 环己烷(95.0) [35 ] 32a NiReO x 2 350 ℃(in⁃situ) 350 0.10 51.8 苯(30.5) [36 ] 33 NiZr⁃I/CMK⁃3 H2 /450 ℃ 300 8 5.00 十六烷 100.0 环己醇(47.9) [37 ] 34c NiCeO2 /rGO H2 /N2 /350 ℃ 300 4 H2 O 23.0 邻苯二酚(9.2) [38 ] 35a NiW/TiO2 H2 S/H2 /400 ℃ 320 5.50 十六烷 100.0 环己烷(65.0) [39 ] 注: a 固定床反应;b 异丙醇作为H供体,p (N2 )=2.00 MPa;c H2 O作为H供体,p (N2 )=10.00 MPa. ...
4
... 除了Ni组分,Mo、Co和Fe等过渡金属也被用来构建愈创木酚加氢反应催化剂,相关研究结果见表2 .Mo是采用较多的催化活性组分,仅次于Ni,负载在碳载体上的Mo基催化剂被广泛用于愈创木酚反应中[5 ,40 ⁃45 ] ,其产物以苯酚或苯为主.S.Ansaloni等[40 ] 在间歇式反应器中测试了Mo负载在SiO2 、Al2 O3 、NaY沸石、MgO、AC和石墨载体上的催化剂的催化活性.结果表明,在350 ℃、4.00 MPa H2 的条件下,负载在AC上的Mo基催化剂对愈创木酚脱甲氧基表现出最佳性能,愈创木酚完全转化,苯酚的收率为72.0%.AC的高表面积使MoO x x 2 C催化剂在愈创木酚的加氢脱氧反应活性[41 ⁃43 ] .Z.Cai等[45 ] 制备了AC负载的MoO2 催化剂,在300 ℃、3.00 MPa H2 的条件下研究了愈创木酚的加氢脱氧反应,得到的主要产物为酚类,且苯酚的收率达到72.0%.上述结果说明,采用以Mo为金属组分、以碳材料为载体的催化剂,产物倾向于芳香族化合物. ...
... [40 ]在间歇式反应器中测试了Mo负载在SiO2 、Al2 O3 、NaY沸石、MgO、AC和石墨载体上的催化剂的催化活性.结果表明,在350 ℃、4.00 MPa H2 的条件下,负载在AC上的Mo基催化剂对愈创木酚脱甲氧基表现出最佳性能,愈创木酚完全转化,苯酚的收率为72.0%.AC的高表面积使MoO x x 2 C催化剂在愈创木酚的加氢脱氧反应活性[41 ⁃43 ] .Z.Cai等[45 ] 制备了AC负载的MoO2 催化剂,在300 ℃、3.00 MPa H2 的条件下研究了愈创木酚的加氢脱氧反应,得到的主要产物为酚类,且苯酚的收率达到72.0%.上述结果说明,采用以Mo为金属组分、以碳材料为载体的催化剂,产物倾向于芳香族化合物. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... 对于串联反应,通过控制反应时间,从动力学上可获得某一中间步骤的产物.在愈创木酚的加氢脱氧反应中,一条典型的反应路径是愈创木酚―苯酚―环己醇―环己烷,随着反应时间的增加,总是由一个产物推进到另一个产物,适当控制反应时间能够提高苯酚、环己醇等中间产物的收率[9 ,25 ,34 ,40 ,45 ] . ...
2
... 除了Ni组分,Mo、Co和Fe等过渡金属也被用来构建愈创木酚加氢反应催化剂,相关研究结果见表2 .Mo是采用较多的催化活性组分,仅次于Ni,负载在碳载体上的Mo基催化剂被广泛用于愈创木酚反应中[5 ,40 ⁃45 ] ,其产物以苯酚或苯为主.S.Ansaloni等[40 ] 在间歇式反应器中测试了Mo负载在SiO2 、Al2 O3 、NaY沸石、MgO、AC和石墨载体上的催化剂的催化活性.结果表明,在350 ℃、4.00 MPa H2 的条件下,负载在AC上的Mo基催化剂对愈创木酚脱甲氧基表现出最佳性能,愈创木酚完全转化,苯酚的收率为72.0%.AC的高表面积使MoO x x 2 C催化剂在愈创木酚的加氢脱氧反应活性[41 ⁃43 ] .Z.Cai等[45 ] 制备了AC负载的MoO2 催化剂,在300 ℃、3.00 MPa H2 的条件下研究了愈创木酚的加氢脱氧反应,得到的主要产物为酚类,且苯酚的收率达到72.0%.上述结果说明,采用以Mo为金属组分、以碳材料为载体的催化剂,产物倾向于芳香族化合物. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
1
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
2
... 除了Ni组分,Mo、Co和Fe等过渡金属也被用来构建愈创木酚加氢反应催化剂,相关研究结果见表2 .Mo是采用较多的催化活性组分,仅次于Ni,负载在碳载体上的Mo基催化剂被广泛用于愈创木酚反应中[5 ,40 ⁃45 ] ,其产物以苯酚或苯为主.S.Ansaloni等[40 ] 在间歇式反应器中测试了Mo负载在SiO2 、Al2 O3 、NaY沸石、MgO、AC和石墨载体上的催化剂的催化活性.结果表明,在350 ℃、4.00 MPa H2 的条件下,负载在AC上的Mo基催化剂对愈创木酚脱甲氧基表现出最佳性能,愈创木酚完全转化,苯酚的收率为72.0%.AC的高表面积使MoO x x 2 C催化剂在愈创木酚的加氢脱氧反应活性[41 ⁃43 ] .Z.Cai等[45 ] 制备了AC负载的MoO2 催化剂,在300 ℃、3.00 MPa H2 的条件下研究了愈创木酚的加氢脱氧反应,得到的主要产物为酚类,且苯酚的收率达到72.0%.上述结果说明,采用以Mo为金属组分、以碳材料为载体的催化剂,产物倾向于芳香族化合物. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
1
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
4
... 除了Ni组分,Mo、Co和Fe等过渡金属也被用来构建愈创木酚加氢反应催化剂,相关研究结果见表2 .Mo是采用较多的催化活性组分,仅次于Ni,负载在碳载体上的Mo基催化剂被广泛用于愈创木酚反应中[5 ,40 ⁃45 ] ,其产物以苯酚或苯为主.S.Ansaloni等[40 ] 在间歇式反应器中测试了Mo负载在SiO2 、Al2 O3 、NaY沸石、MgO、AC和石墨载体上的催化剂的催化活性.结果表明,在350 ℃、4.00 MPa H2 的条件下,负载在AC上的Mo基催化剂对愈创木酚脱甲氧基表现出最佳性能,愈创木酚完全转化,苯酚的收率为72.0%.AC的高表面积使MoO x x 2 C催化剂在愈创木酚的加氢脱氧反应活性[41 ⁃43 ] .Z.Cai等[45 ] 制备了AC负载的MoO2 催化剂,在300 ℃、3.00 MPa H2 的条件下研究了愈创木酚的加氢脱氧反应,得到的主要产物为酚类,且苯酚的收率达到72.0%.上述结果说明,采用以Mo为金属组分、以碳材料为载体的催化剂,产物倾向于芳香族化合物. ...
... [45 ]制备了AC负载的MoO2 催化剂,在300 ℃、3.00 MPa H2 的条件下研究了愈创木酚的加氢脱氧反应,得到的主要产物为酚类,且苯酚的收率达到72.0%.上述结果说明,采用以Mo为金属组分、以碳材料为载体的催化剂,产物倾向于芳香族化合物. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... 对于串联反应,通过控制反应时间,从动力学上可获得某一中间步骤的产物.在愈创木酚的加氢脱氧反应中,一条典型的反应路径是愈创木酚―苯酚―环己醇―环己烷,随着反应时间的增加,总是由一个产物推进到另一个产物,适当控制反应时间能够提高苯酚、环己醇等中间产物的收率[9 ,25 ,34 ,40 ,45 ] . ...
2
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
2
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
4
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... [
48 ]
13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... 载体除了起到担载和分散活性组分的作用,还可能影响催化功能.多种类型的碳材料被广泛用作载体,例如AC、CNT和石墨烯等.相较于酸性较高的分子筛、金属氧化物载体,碳材料由于普遍缺乏酸位点,可以在一定程度上抑制焦炭形成,提高催化剂的寿命[5 ⁃6 ] .但是,由于碳基载体的酸度太低,在活化含氧基团方面显得能力较低,因此基于碳材料的催化剂通常不是依靠载体构建酸催化功能.例如,Ni5 ⁃Fe1 /CNT[32 ] 、Co⁃MoO2 /C[48 ] 、RuRe/C[61 ] 等碳基催化剂采用双金属组分,其中呈氧化态的金属助剂发挥酸催化功能.对SiO2 、Al2 O3 和分子筛等具有一定酸性的载体,酸性太弱的载体会体现酸功能不足,中度酸性载体构造的催化剂往往具有解离CAR -OCH3 的能力,可提高对苯酚的选择性[20 ⁃23 ] ,而基于强酸性载体的催化剂有可能使苯酚的羟基也脱除,得到完全脱氧产物. ...
1
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
2
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
2
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... R.N.Olcese等[52 ] 探索了Fe/SiO2 对愈创木酚的加氢脱氧反应,发现负载15.0%的Fe催化剂在愈创木酚的加氢脱氧中对苯环的加氢反应具有惰性,可以将反应控制在产物为芳香族化合物,从而防止愈创木酚过度加氢.基于Fe/CeO2 [51 ] 和Fe/AC[21 ] 的反应,产物主要也是芳香族化合物,可见基于Fe催化剂可以选择性催化芳基醚键的断裂,有利于生成芳香类产物.然而,Fe基催化剂的催化活性较低,需要在300~400 ℃的高温条件下才能转化.为了克服这一缺陷,通常引入另一种金属,以显著提高催化性能[32 ,53 ⁃54 ] .例如,N.T.T.Tran等[53 ] 以Pd⁃Fe/Al⁃MCM⁃41为催化剂将愈创木酚转化为苯酚,其收率约为50.0%. ...
2
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Mo/AC 350 1.0 4.00 十二烷 99.9 苯酚(72.0) [40 ] 2 Mo2 C 350 5.00 十二烷 30.0 苯酚(20.0) [5 ] 3 Mo2 C/CNF 350 4.0 2.00 正十烷 80.0 苯酚(38.0) [41 ] 4 Mo2 C/CNF H2 /350 ℃ 300 2.0 2.00 正十烷 67.0 含一个O产物(27.0) [42 ] 5 β⁃Mo2 C/CNF 300 2.0 2.00 正十烷 67.0 苯酚(28.0) [43 ] 6 MoWC H2 /450 ℃ 350 1.0 2.75 正十烷 93.0 苯(65.0) [44 ] 7 MoO2 /AC H2 /600 ℃ 300 3.0 3.00 四氢萘 98.0 苯酚(72.0) [45 ] 8 Co/ZrP H2 /500 ℃ 300 2.5 4.00 十二烷 100.0 环己烷(76.0) [46 ] 9 Co/HMETS⁃10 H2 /400 ℃ 280 2.0 2.00 十二烷 99.5 环己烷(96.9) [47 ] 10 Co/G H2 /300 ℃ 300 4.0 5.00 十二烷 77.0 苯酚(52.5) [24 ] 11 Co⁃MoO2 /C 340 4.0 0.80 正己烷 97.0 苯+甲苯(61.1) [48 ] 12 Co⁃MoO2 /C 340 4.0 0.80 乙醇 84.5 苯酚+邻苯二酚+甲苯(75.2) [48 ] 13 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 350 2.0 8.00 十六烷 100.0 苯(70.0) [49 ] 14 CoMoS/γ⁃Al2 O3 H2 /H2 S/400 ℃ 250 5.50 二甲苯+乙苯 100.0 苯酚(55.0) [50 ] 15 Fe/CeO2 400 2.0~4.0 0.10 99.0 苯酚(56.0) [51 ] 16 Fe/AC H2 /Ar/550 ℃ 300 0.10 87.0 1,2⁃二甲氧基苯(82.0) [21 ] 17 Fe/SiO2 H2 /500 ℃ 400 0.10 74.0 苯+苯酚+苯甲醚(38.0) [52 ] Co基催化剂对产物的选择性因载体而变化.Co负载在磷酸锆(Co/ZrP)[46 ] 和分子筛(Co/HMETS⁃10)[47 ] 的催化剂获得的产物以环己烷为主,而Co负载在石墨碳(Co/G)[24 ] 的催化剂会得到52.5%的苯酚产物.Co与Mo结合构建的双金属催化剂对愈创木酚加氢反应活性很高,产物以芳香族化合物为主[48 ⁃50 ] ,其中CoMoS/γ⁃Al2 O3 催化剂获得了70.0%的苯收率.预硫化处理后催化剂表面含硫基团,亲核SH-基团与模型化合物相互作用促进了催化反应.然而,这些催化剂需要持续引入S来保持MoS2 催化剂的活性,不可避免地污染加氢脱氧产物,加上γ⁃Al2 O3 的强酸性很容易使催化剂积碳,催化剂容易失活. ...
... R.N.Olcese等[52 ] 探索了Fe/SiO2 对愈创木酚的加氢脱氧反应,发现负载15.0%的Fe催化剂在愈创木酚的加氢脱氧中对苯环的加氢反应具有惰性,可以将反应控制在产物为芳香族化合物,从而防止愈创木酚过度加氢.基于Fe/CeO2 [51 ] 和Fe/AC[21 ] 的反应,产物主要也是芳香族化合物,可见基于Fe催化剂可以选择性催化芳基醚键的断裂,有利于生成芳香类产物.然而,Fe基催化剂的催化活性较低,需要在300~400 ℃的高温条件下才能转化.为了克服这一缺陷,通常引入另一种金属,以显著提高催化性能[32 ,53 ⁃54 ] .例如,N.T.T.Tran等[53 ] 以Pd⁃Fe/Al⁃MCM⁃41为催化剂将愈创木酚转化为苯酚,其收率约为50.0%. ...
5
... R.N.Olcese等[52 ] 探索了Fe/SiO2 对愈创木酚的加氢脱氧反应,发现负载15.0%的Fe催化剂在愈创木酚的加氢脱氧中对苯环的加氢反应具有惰性,可以将反应控制在产物为芳香族化合物,从而防止愈创木酚过度加氢.基于Fe/CeO2 [51 ] 和Fe/AC[21 ] 的反应,产物主要也是芳香族化合物,可见基于Fe催化剂可以选择性催化芳基醚键的断裂,有利于生成芳香类产物.然而,Fe基催化剂的催化活性较低,需要在300~400 ℃的高温条件下才能转化.为了克服这一缺陷,通常引入另一种金属,以显著提高催化性能[32 ,53 ⁃54 ] .例如,N.T.T.Tran等[53 ] 以Pd⁃Fe/Al⁃MCM⁃41为催化剂将愈创木酚转化为苯酚,其收率约为50.0%. ...
... [53 ]以Pd⁃Fe/Al⁃MCM⁃41为催化剂将愈创木酚转化为苯酚,其收率约为50.0%. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
... 单金属Pd催化剂[73 ] 及PdNi[10 ] 、PdFe[53 ⁃54 ] 、PdRe[74 ] 双金属催化剂也被用于愈创木酚的加氢脱氧反应.基于Pd催化剂的产物种类也很多,有的以芳香族化合物为主[10 ,53 ,74 ] ,有的是芳环加氢饱和的产物[73 ] .H.Shafaghat等[73 ] 以Pd/C为催化剂,探究了醇类(甲醇、乙醇、1⁃丙醇、1⁃丁醇、异丙醇和2⁃丁醇等)替代H2 的可行性.结果表明,只有仲醇可以作为Pd/C上愈创木酚转化的H供体.同时,当使用异丙醇作为H供体时,生成较多的1⁃甲丙基环己烷(收率为49.3%).S.T.Thompson等[74 ] 采用Re对Pd/C催化剂进行修饰,结果表明,在400 ℃原位还原后,PdRe/C可获得完全脱氧产物(苯和环己烷),其中苯的收率为35.0%.PdRe双金属催化剂结合了Pd/C的脱甲氧基和加氢功能与Re/C的对苯酚的脱氧功能. ...
... ,53 ,74 ],有的是芳环加氢饱和的产物[73 ] .H.Shafaghat等[73 ] 以Pd/C为催化剂,探究了醇类(甲醇、乙醇、1⁃丙醇、1⁃丁醇、异丙醇和2⁃丁醇等)替代H2 的可行性.结果表明,只有仲醇可以作为Pd/C上愈创木酚转化的H供体.同时,当使用异丙醇作为H供体时,生成较多的1⁃甲丙基环己烷(收率为49.3%).S.T.Thompson等[74 ] 采用Re对Pd/C催化剂进行修饰,结果表明,在400 ℃原位还原后,PdRe/C可获得完全脱氧产物(苯和环己烷),其中苯的收率为35.0%.PdRe双金属催化剂结合了Pd/C的脱甲氧基和加氢功能与Re/C的对苯酚的脱氧功能. ...
3
... R.N.Olcese等[52 ] 探索了Fe/SiO2 对愈创木酚的加氢脱氧反应,发现负载15.0%的Fe催化剂在愈创木酚的加氢脱氧中对苯环的加氢反应具有惰性,可以将反应控制在产物为芳香族化合物,从而防止愈创木酚过度加氢.基于Fe/CeO2 [51 ] 和Fe/AC[21 ] 的反应,产物主要也是芳香族化合物,可见基于Fe催化剂可以选择性催化芳基醚键的断裂,有利于生成芳香类产物.然而,Fe基催化剂的催化活性较低,需要在300~400 ℃的高温条件下才能转化.为了克服这一缺陷,通常引入另一种金属,以显著提高催化性能[32 ,53 ⁃54 ] .例如,N.T.T.Tran等[53 ] 以Pd⁃Fe/Al⁃MCM⁃41为催化剂将愈创木酚转化为苯酚,其收率约为50.0%. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
... 单金属Pd催化剂[73 ] 及PdNi[10 ] 、PdFe[53 ⁃54 ] 、PdRe[74 ] 双金属催化剂也被用于愈创木酚的加氢脱氧反应.基于Pd催化剂的产物种类也很多,有的以芳香族化合物为主[10 ,53 ,74 ] ,有的是芳环加氢饱和的产物[73 ] .H.Shafaghat等[73 ] 以Pd/C为催化剂,探究了醇类(甲醇、乙醇、1⁃丙醇、1⁃丁醇、异丙醇和2⁃丁醇等)替代H2 的可行性.结果表明,只有仲醇可以作为Pd/C上愈创木酚转化的H供体.同时,当使用异丙醇作为H供体时,生成较多的1⁃甲丙基环己烷(收率为49.3%).S.T.Thompson等[74 ] 采用Re对Pd/C催化剂进行修饰,结果表明,在400 ℃原位还原后,PdRe/C可获得完全脱氧产物(苯和环己烷),其中苯的收率为35.0%.PdRe双金属催化剂结合了Pd/C的脱甲氧基和加氢功能与Re/C的对苯酚的脱氧功能. ...
6
... Ru基催化剂因其较高的催化活性在加氢脱氧反应中被广泛研究(见表3 ).从表3 可以看出,Ru负载在氧化物[55 ⁃57 ] 、分子筛[58 ⁃60 ] 和碳材料[61 ⁃64 ] 上的催化剂以及Ru与过渡金属复合催化剂[65 ⁃68 ] 在240 ℃左右的温度下均能催化愈创木酚加氢反应,其中Ru/TiO2 [55 ] 、Ru/C[61 ] 和Ru⁃MnO/CNTs[68 ] 催化剂获得产物以环己醇为主,Ru/Cl/TiO2 催化剂[55 ] 和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... [55 ]、Ru/C[61 ] 和Ru⁃MnO/CNTs[68 ] 催化剂获得产物以环己醇为主,Ru/Cl/TiO2 催化剂[55 ] 和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... [55 ]和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... [55 ]的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
... [
55 ]
3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
1
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
2
... Ru基催化剂因其较高的催化活性在加氢脱氧反应中被广泛研究(见表3 ).从表3 可以看出,Ru负载在氧化物[55 ⁃57 ] 、分子筛[58 ⁃60 ] 和碳材料[61 ⁃64 ] 上的催化剂以及Ru与过渡金属复合催化剂[65 ⁃68 ] 在240 ℃左右的温度下均能催化愈创木酚加氢反应,其中Ru/TiO2 [55 ] 、Ru/C[61 ] 和Ru⁃MnO/CNTs[68 ] 催化剂获得产物以环己醇为主,Ru/Cl/TiO2 催化剂[55 ] 和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
2
... Ru基催化剂因其较高的催化活性在加氢脱氧反应中被广泛研究(见表3 ).从表3 可以看出,Ru负载在氧化物[55 ⁃57 ] 、分子筛[58 ⁃60 ] 和碳材料[61 ⁃64 ] 上的催化剂以及Ru与过渡金属复合催化剂[65 ⁃68 ] 在240 ℃左右的温度下均能催化愈创木酚加氢反应,其中Ru/TiO2 [55 ] 、Ru/C[61 ] 和Ru⁃MnO/CNTs[68 ] 催化剂获得产物以环己醇为主,Ru/Cl/TiO2 催化剂[55 ] 和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
2
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
... [
59 ]
8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
2
... Ru基催化剂因其较高的催化活性在加氢脱氧反应中被广泛研究(见表3 ).从表3 可以看出,Ru负载在氧化物[55 ⁃57 ] 、分子筛[58 ⁃60 ] 和碳材料[61 ⁃64 ] 上的催化剂以及Ru与过渡金属复合催化剂[65 ⁃68 ] 在240 ℃左右的温度下均能催化愈创木酚加氢反应,其中Ru/TiO2 [55 ] 、Ru/C[61 ] 和Ru⁃MnO/CNTs[68 ] 催化剂获得产物以环己醇为主,Ru/Cl/TiO2 催化剂[55 ] 和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
7
... Ru基催化剂因其较高的催化活性在加氢脱氧反应中被广泛研究(见表3 ).从表3 可以看出,Ru负载在氧化物[55 ⁃57 ] 、分子筛[58 ⁃60 ] 和碳材料[61 ⁃64 ] 上的催化剂以及Ru与过渡金属复合催化剂[65 ⁃68 ] 在240 ℃左右的温度下均能催化愈创木酚加氢反应,其中Ru/TiO2 [55 ] 、Ru/C[61 ] 和Ru⁃MnO/CNTs[68 ] 催化剂获得产物以环己醇为主,Ru/Cl/TiO2 催化剂[55 ] 和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... [61 ]和Ru⁃MnO/CNTs[68 ] 催化剂获得产物以环己醇为主,Ru/Cl/TiO2 催化剂[55 ] 和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
... [
61 ]
注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
... 由于两种金属之间的协同作用可能产生新的催化功能,一些研究者为愈创木酚的加氢脱氧反应开发了Ru和Re[61 ] 、Co[64 ⁃66 ] 、Ni[67 ] 、Mn[68 ] 等组分构成的双金属催化剂.M.Kim等[61 ] 以异丙醇为H供体进行了催化愈创木酚的氢转移反应.结果表明,愈创木酚在Ru/C催化剂上主要转化为部分脱氧的环己醇产物,其收率为70.0%;在Ru/C催化剂中加入Re显著提高了C-O氢解的速率,导致完全脱氧的环己烷收率(约56.0%)显著提高.RuRe/C双金属催化剂超强的脱氧能力归因于其双功能特性,即Ru催化了愈创木酚的加氢和部分氢解反应,Re提供的酸位促进了C-O的氢解.R.X.Li等[67 ] 通过与单金属催化剂的比较,在260 ℃、1.00 MPa H2 的条件下,研究了双金属RuNi/SiO2 ⁃ZrO2 催化剂的吸附性能和加氢脱氧性能之间的促进关系.RuNi双金属催化剂对愈创木酚的加氢脱氧的催化性能比Ru或Ni单金属催化剂优异,环己烷的收率达到74.5%.该课题组在此基础上还研究了RuCo/SiO2 ⁃ZrO2 双金属催化剂对愈创木酚的加氢脱氧反应性能[66 ] .结果表明,催化剂中的两种金属组分之间有很强的相互作用,电子从Ru转移到过渡金属,从而导致与Ru单金属催化剂不同的催化性能. ...
... [61 ]以异丙醇为H供体进行了催化愈创木酚的氢转移反应.结果表明,愈创木酚在Ru/C催化剂上主要转化为部分脱氧的环己醇产物,其收率为70.0%;在Ru/C催化剂中加入Re显著提高了C-O氢解的速率,导致完全脱氧的环己烷收率(约56.0%)显著提高.RuRe/C双金属催化剂超强的脱氧能力归因于其双功能特性,即Ru催化了愈创木酚的加氢和部分氢解反应,Re提供的酸位促进了C-O的氢解.R.X.Li等[67 ] 通过与单金属催化剂的比较,在260 ℃、1.00 MPa H2 的条件下,研究了双金属RuNi/SiO2 ⁃ZrO2 催化剂的吸附性能和加氢脱氧性能之间的促进关系.RuNi双金属催化剂对愈创木酚的加氢脱氧的催化性能比Ru或Ni单金属催化剂优异,环己烷的收率达到74.5%.该课题组在此基础上还研究了RuCo/SiO2 ⁃ZrO2 双金属催化剂对愈创木酚的加氢脱氧反应性能[66 ] .结果表明,催化剂中的两种金属组分之间有很强的相互作用,电子从Ru转移到过渡金属,从而导致与Ru单金属催化剂不同的催化性能. ...
... 载体除了起到担载和分散活性组分的作用,还可能影响催化功能.多种类型的碳材料被广泛用作载体,例如AC、CNT和石墨烯等.相较于酸性较高的分子筛、金属氧化物载体,碳材料由于普遍缺乏酸位点,可以在一定程度上抑制焦炭形成,提高催化剂的寿命[5 ⁃6 ] .但是,由于碳基载体的酸度太低,在活化含氧基团方面显得能力较低,因此基于碳材料的催化剂通常不是依靠载体构建酸催化功能.例如,Ni5 ⁃Fe1 /CNT[32 ] 、Co⁃MoO2 /C[48 ] 、RuRe/C[61 ] 等碳基催化剂采用双金属组分,其中呈氧化态的金属助剂发挥酸催化功能.对SiO2 、Al2 O3 和分子筛等具有一定酸性的载体,酸性太弱的载体会体现酸功能不足,中度酸性载体构造的催化剂往往具有解离CAR -OCH3 的能力,可提高对苯酚的选择性[20 ⁃23 ] ,而基于强酸性载体的催化剂有可能使苯酚的羟基也脱除,得到完全脱氧产物. ...
2
... Ru基催化剂因其较高的催化活性在加氢脱氧反应中被广泛研究(见表3 ).从表3 可以看出,Ru负载在氧化物[55 ⁃57 ] 、分子筛[58 ⁃60 ] 和碳材料[61 ⁃64 ] 上的催化剂以及Ru与过渡金属复合催化剂[65 ⁃68 ] 在240 ℃左右的温度下均能催化愈创木酚加氢反应,其中Ru/TiO2 [55 ] 、Ru/C[61 ] 和Ru⁃MnO/CNTs[68 ] 催化剂获得产物以环己醇为主,Ru/Cl/TiO2 催化剂[55 ] 和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
1
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
3
... Ru基催化剂因其较高的催化活性在加氢脱氧反应中被广泛研究(见表3 ).从表3 可以看出,Ru负载在氧化物[55 ⁃57 ] 、分子筛[58 ⁃60 ] 和碳材料[61 ⁃64 ] 上的催化剂以及Ru与过渡金属复合催化剂[65 ⁃68 ] 在240 ℃左右的温度下均能催化愈创木酚加氢反应,其中Ru/TiO2 [55 ] 、Ru/C[61 ] 和Ru⁃MnO/CNTs[68 ] 催化剂获得产物以环己醇为主,Ru/Cl/TiO2 催化剂[55 ] 和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
... 由于两种金属之间的协同作用可能产生新的催化功能,一些研究者为愈创木酚的加氢脱氧反应开发了Ru和Re[61 ] 、Co[64 ⁃66 ] 、Ni[67 ] 、Mn[68 ] 等组分构成的双金属催化剂.M.Kim等[61 ] 以异丙醇为H供体进行了催化愈创木酚的氢转移反应.结果表明,愈创木酚在Ru/C催化剂上主要转化为部分脱氧的环己醇产物,其收率为70.0%;在Ru/C催化剂中加入Re显著提高了C-O氢解的速率,导致完全脱氧的环己烷收率(约56.0%)显著提高.RuRe/C双金属催化剂超强的脱氧能力归因于其双功能特性,即Ru催化了愈创木酚的加氢和部分氢解反应,Re提供的酸位促进了C-O的氢解.R.X.Li等[67 ] 通过与单金属催化剂的比较,在260 ℃、1.00 MPa H2 的条件下,研究了双金属RuNi/SiO2 ⁃ZrO2 催化剂的吸附性能和加氢脱氧性能之间的促进关系.RuNi双金属催化剂对愈创木酚的加氢脱氧的催化性能比Ru或Ni单金属催化剂优异,环己烷的收率达到74.5%.该课题组在此基础上还研究了RuCo/SiO2 ⁃ZrO2 双金属催化剂对愈创木酚的加氢脱氧反应性能[66 ] .结果表明,催化剂中的两种金属组分之间有很强的相互作用,电子从Ru转移到过渡金属,从而导致与Ru单金属催化剂不同的催化性能. ...
4
... Ru基催化剂因其较高的催化活性在加氢脱氧反应中被广泛研究(见表3 ).从表3 可以看出,Ru负载在氧化物[55 ⁃57 ] 、分子筛[58 ⁃60 ] 和碳材料[61 ⁃64 ] 上的催化剂以及Ru与过渡金属复合催化剂[65 ⁃68 ] 在240 ℃左右的温度下均能催化愈创木酚加氢反应,其中Ru/TiO2 [55 ] 、Ru/C[61 ] 和Ru⁃MnO/CNTs[68 ] 催化剂获得产物以环己醇为主,Ru/Cl/TiO2 催化剂[55 ] 和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
... 在愈创木酚的加氢脱氧反应中,大部分研究人员选择H2 作为H供体.H2 是反应物,氢分压的增加提高H2 在溶液中的溶解度,进而增加催化剂活性位上活性氢的浓度.研究表明[8 ,65 ] ,氢分压不仅影响加氢程度,随着反应的进行,更倾向于沿着愈创木酚―苯酚―环己醇―环己烷的方向递进,而且还会改变反应路径.M.H.Zhou等[8 ] 基于NiCo/γ⁃Al2 O3 催化剂研究了初始氢分压的影响.结果表明,随着初始氢分压增加,环己醇选择性逐渐降低,同时1⁃甲基⁃1,2⁃环己二醇(反式和顺式)的总选择性增加.苯环加氢反应需要较多的活性氢,所以当氢分压增加时,愈创木酚更容易通过加氢苯环生成2⁃甲氧基环己醇,进而异构化生成1⁃甲基⁃1,2⁃环己二醇(反式和顺式).该反应路线消耗较多的愈创木酚,使愈创木酚去甲氧基生成苯酚,苯酚再进一步加氢生成环己醇的反应被削弱.Q.Q.Xu等[65 ] 利用Ru⁃Co/C催化剂探究氢分压(0.50~2.00 MPa)对愈创木酚加氢脱氧反应(200 ℃、1.5 h)的影响.结果表明,在0.5 MPa的氢分压下,生成了大量的苯酚(收率为29.4%).因此,较低的氢分压能够降低苯环饱和反应的可能性,提高苯酚选择性. ...
... [65 ]利用Ru⁃Co/C催化剂探究氢分压(0.50~2.00 MPa)对愈创木酚加氢脱氧反应(200 ℃、1.5 h)的影响.结果表明,在0.5 MPa的氢分压下,生成了大量的苯酚(收率为29.4%).因此,较低的氢分压能够降低苯环饱和反应的可能性,提高苯酚选择性. ...
3
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
... 由于两种金属之间的协同作用可能产生新的催化功能,一些研究者为愈创木酚的加氢脱氧反应开发了Ru和Re[61 ] 、Co[64 ⁃66 ] 、Ni[67 ] 、Mn[68 ] 等组分构成的双金属催化剂.M.Kim等[61 ] 以异丙醇为H供体进行了催化愈创木酚的氢转移反应.结果表明,愈创木酚在Ru/C催化剂上主要转化为部分脱氧的环己醇产物,其收率为70.0%;在Ru/C催化剂中加入Re显著提高了C-O氢解的速率,导致完全脱氧的环己烷收率(约56.0%)显著提高.RuRe/C双金属催化剂超强的脱氧能力归因于其双功能特性,即Ru催化了愈创木酚的加氢和部分氢解反应,Re提供的酸位促进了C-O的氢解.R.X.Li等[67 ] 通过与单金属催化剂的比较,在260 ℃、1.00 MPa H2 的条件下,研究了双金属RuNi/SiO2 ⁃ZrO2 催化剂的吸附性能和加氢脱氧性能之间的促进关系.RuNi双金属催化剂对愈创木酚的加氢脱氧的催化性能比Ru或Ni单金属催化剂优异,环己烷的收率达到74.5%.该课题组在此基础上还研究了RuCo/SiO2 ⁃ZrO2 双金属催化剂对愈创木酚的加氢脱氧反应性能[66 ] .结果表明,催化剂中的两种金属组分之间有很强的相互作用,电子从Ru转移到过渡金属,从而导致与Ru单金属催化剂不同的催化性能. ...
... [66 ].结果表明,催化剂中的两种金属组分之间有很强的相互作用,电子从Ru转移到过渡金属,从而导致与Ru单金属催化剂不同的催化性能. ...
3
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
... 由于两种金属之间的协同作用可能产生新的催化功能,一些研究者为愈创木酚的加氢脱氧反应开发了Ru和Re[61 ] 、Co[64 ⁃66 ] 、Ni[67 ] 、Mn[68 ] 等组分构成的双金属催化剂.M.Kim等[61 ] 以异丙醇为H供体进行了催化愈创木酚的氢转移反应.结果表明,愈创木酚在Ru/C催化剂上主要转化为部分脱氧的环己醇产物,其收率为70.0%;在Ru/C催化剂中加入Re显著提高了C-O氢解的速率,导致完全脱氧的环己烷收率(约56.0%)显著提高.RuRe/C双金属催化剂超强的脱氧能力归因于其双功能特性,即Ru催化了愈创木酚的加氢和部分氢解反应,Re提供的酸位促进了C-O的氢解.R.X.Li等[67 ] 通过与单金属催化剂的比较,在260 ℃、1.00 MPa H2 的条件下,研究了双金属RuNi/SiO2 ⁃ZrO2 催化剂的吸附性能和加氢脱氧性能之间的促进关系.RuNi双金属催化剂对愈创木酚的加氢脱氧的催化性能比Ru或Ni单金属催化剂优异,环己烷的收率达到74.5%.该课题组在此基础上还研究了RuCo/SiO2 ⁃ZrO2 双金属催化剂对愈创木酚的加氢脱氧反应性能[66 ] .结果表明,催化剂中的两种金属组分之间有很强的相互作用,电子从Ru转移到过渡金属,从而导致与Ru单金属催化剂不同的催化性能. ...
... [67 ]通过与单金属催化剂的比较,在260 ℃、1.00 MPa H2 的条件下,研究了双金属RuNi/SiO2 ⁃ZrO2 催化剂的吸附性能和加氢脱氧性能之间的促进关系.RuNi双金属催化剂对愈创木酚的加氢脱氧的催化性能比Ru或Ni单金属催化剂优异,环己烷的收率达到74.5%.该课题组在此基础上还研究了RuCo/SiO2 ⁃ZrO2 双金属催化剂对愈创木酚的加氢脱氧反应性能[66 ] .结果表明,催化剂中的两种金属组分之间有很强的相互作用,电子从Ru转移到过渡金属,从而导致与Ru单金属催化剂不同的催化性能. ...
4
... Ru基催化剂因其较高的催化活性在加氢脱氧反应中被广泛研究(见表3 ).从表3 可以看出,Ru负载在氧化物[55 ⁃57 ] 、分子筛[58 ⁃60 ] 和碳材料[61 ⁃64 ] 上的催化剂以及Ru与过渡金属复合催化剂[65 ⁃68 ] 在240 ℃左右的温度下均能催化愈创木酚加氢反应,其中Ru/TiO2 [55 ] 、Ru/C[61 ] 和Ru⁃MnO/CNTs[68 ] 催化剂获得产物以环己醇为主,Ru/Cl/TiO2 催化剂[55 ] 和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... [68 ]催化剂获得产物以环己醇为主,Ru/Cl/TiO2 催化剂[55 ] 和Ru/AC催化剂[62 ] 获得酚类产物,而其他催化剂获得的主产物大多为环己烷.由此可见,Ru基催化剂更容易使愈创木酚的加氢脱氧反应生成苯环饱和的产物,Ru组分对苯环加氢有很高的催化活性.X.C.Wang等[55 ] 的研究结果表明,负载于TiO2 上的Ru催化剂通过Cl改性能调变其催化性能.与未改性的Ru/TiO2 催化剂相比,Ru/Cl/TiO2 表现出适度的去甲氧基化的催化活性,获得更高的苯酚选择性.Cl组分很可能定位于金属⁃载体界面和金属颗粒表面,对Ru和Ti组分起到电子修饰作用,使富含电子的Ru抑制苯环加氢反应,而且TiO2 表面缺陷促进脱氧反应. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Ru/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 71.7 环己醇(51.0) [55 ] 2 Ru/Cl/TiO2 H2 /250 ℃ 240 1.00 1.00 二氧六环 45.0 苯酚(29.0) [55 ] 3 Ru/WZrO2 H2 /Ar/400 ℃ 270 1.00 4.00 H2 O 96.8 不含O产物(56.5) [56 ] 4 Ru/TiO2 ⁃γAl2 O3 240 6.00 1.00 辛烷 91.4 环己烷(81.7) [57 ] 5 Ru/Al⁃HMS(10) H2 /400 ℃ 250 3.00 5.00 甲醇 97.0 环己烷(67.0) [58 ] 6 Ru/BEA⁃12.5 250 2.00 4.00 100.0 环己烷(71.9) [59 ] 7 Ru/Al2 O3 250 2.00 4.00 100.0 2⁃甲氧基环己醇(74.8) [59 ] 8 Ru/Hβ⁃25 180 2.00 2.00 正己烷 100.0 环己烷(100.0) [60 ] 9a Ru/C H2 /250 ℃ 200 5.00 异丙醇 99.0 环己醇(70.0) [61 ] 10 Ru/AC 250 4.00 5.00 H2 O 25.1 苯酚+邻苯二酚 [62 ] 11 Ru/C+H2 WO4 260 4.00 3.00 辛烷 99.9 环己烷(99.9) [63 ] 12 RuCo⁃300 H2 /Ar/300 ℃ 310 1.00 正十烷 99.0 环己烷(85.3) [64 ] 13 RuCo/C⁃600 H2 /600 ℃ 200 1.50 1.00 正十烷 100.0 环己烷(94.0) [65 ] 14 RuCo/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 99.9 环己烷(88.3) [66 ] 15 RuNi/SiO2 ⁃ZrO2 H2 /550 ℃ 260 4.00 1.00 辛烷 100.0 环己烷(74.5) [67 ] 16 Ru⁃MnO/CNTs H2 /400 ℃ 200 3.33 2.00 十氢萘 99.4 环己醇(85.8) [68 ] 17 a RuRe/C H2 /250 ℃ 200 5.00 异丙醇 95.0 环己烷(56.0) [61 ] 注: a 异丙醇作为H供体,p (N2 )=2.00 MPa. ...
... 由于两种金属之间的协同作用可能产生新的催化功能,一些研究者为愈创木酚的加氢脱氧反应开发了Ru和Re[61 ] 、Co[64 ⁃66 ] 、Ni[67 ] 、Mn[68 ] 等组分构成的双金属催化剂.M.Kim等[61 ] 以异丙醇为H供体进行了催化愈创木酚的氢转移反应.结果表明,愈创木酚在Ru/C催化剂上主要转化为部分脱氧的环己醇产物,其收率为70.0%;在Ru/C催化剂中加入Re显著提高了C-O氢解的速率,导致完全脱氧的环己烷收率(约56.0%)显著提高.RuRe/C双金属催化剂超强的脱氧能力归因于其双功能特性,即Ru催化了愈创木酚的加氢和部分氢解反应,Re提供的酸位促进了C-O的氢解.R.X.Li等[67 ] 通过与单金属催化剂的比较,在260 ℃、1.00 MPa H2 的条件下,研究了双金属RuNi/SiO2 ⁃ZrO2 催化剂的吸附性能和加氢脱氧性能之间的促进关系.RuNi双金属催化剂对愈创木酚的加氢脱氧的催化性能比Ru或Ni单金属催化剂优异,环己烷的收率达到74.5%.该课题组在此基础上还研究了RuCo/SiO2 ⁃ZrO2 双金属催化剂对愈创木酚的加氢脱氧反应性能[66 ] .结果表明,催化剂中的两种金属组分之间有很强的相互作用,电子从Ru转移到过渡金属,从而导致与Ru单金属催化剂不同的催化性能. ...
2
... 表4 为用于愈创木酚加氢脱氧反应的其他贵金属催化剂.E.H.Lee等[69 ] 在HZSM⁃5、Si⁃MCM⁃48、Mesoβ和Hβ等不同载体负载贵金属Pt,研究了载体对愈创木酚加氢脱氧反应的影响.结果表明,较大的孔隙结构和较多的酸位点是愈创木酚转化的必要条件;Pt负载在小孔径的HZSM⁃5和没有酸位点的Si⁃MCM⁃48上,愈创木酚的转化率较低;Pt/Mesoβ和Pt/Hβ同时具有大孔和强酸位点,愈创木酚转化率大于90.0%,产物以环己烷为主.Pt/Hβ催化剂在较低的氢分压(0.10 MPa)下产生较多的芳香族化合物[70 ] .Pt/Nb2 O5 催化剂在0.1 MPa的氢分压下也获得苯酚产物[71 ] .由此可见,采用较低氢分压能够抑制苯环加氢反应,有利于生成苯酚等芳香族化合物. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
2
... 表4 为用于愈创木酚加氢脱氧反应的其他贵金属催化剂.E.H.Lee等[69 ] 在HZSM⁃5、Si⁃MCM⁃48、Mesoβ和Hβ等不同载体负载贵金属Pt,研究了载体对愈创木酚加氢脱氧反应的影响.结果表明,较大的孔隙结构和较多的酸位点是愈创木酚转化的必要条件;Pt负载在小孔径的HZSM⁃5和没有酸位点的Si⁃MCM⁃48上,愈创木酚的转化率较低;Pt/Mesoβ和Pt/Hβ同时具有大孔和强酸位点,愈创木酚转化率大于90.0%,产物以环己烷为主.Pt/Hβ催化剂在较低的氢分压(0.10 MPa)下产生较多的芳香族化合物[70 ] .Pt/Nb2 O5 催化剂在0.1 MPa的氢分压下也获得苯酚产物[71 ] .由此可见,采用较低氢分压能够抑制苯环加氢反应,有利于生成苯酚等芳香族化合物. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
2
... 表4 为用于愈创木酚加氢脱氧反应的其他贵金属催化剂.E.H.Lee等[69 ] 在HZSM⁃5、Si⁃MCM⁃48、Mesoβ和Hβ等不同载体负载贵金属Pt,研究了载体对愈创木酚加氢脱氧反应的影响.结果表明,较大的孔隙结构和较多的酸位点是愈创木酚转化的必要条件;Pt负载在小孔径的HZSM⁃5和没有酸位点的Si⁃MCM⁃48上,愈创木酚的转化率较低;Pt/Mesoβ和Pt/Hβ同时具有大孔和强酸位点,愈创木酚转化率大于90.0%,产物以环己烷为主.Pt/Hβ催化剂在较低的氢分压(0.10 MPa)下产生较多的芳香族化合物[70 ] .Pt/Nb2 O5 催化剂在0.1 MPa的氢分压下也获得苯酚产物[71 ] .由此可见,采用较低氢分压能够抑制苯环加氢反应,有利于生成苯酚等芳香族化合物. ...
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
1
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
4
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
... 单金属Pd催化剂[73 ] 及PdNi[10 ] 、PdFe[53 ⁃54 ] 、PdRe[74 ] 双金属催化剂也被用于愈创木酚的加氢脱氧反应.基于Pd催化剂的产物种类也很多,有的以芳香族化合物为主[10 ,53 ,74 ] ,有的是芳环加氢饱和的产物[73 ] .H.Shafaghat等[73 ] 以Pd/C为催化剂,探究了醇类(甲醇、乙醇、1⁃丙醇、1⁃丁醇、异丙醇和2⁃丁醇等)替代H2 的可行性.结果表明,只有仲醇可以作为Pd/C上愈创木酚转化的H供体.同时,当使用异丙醇作为H供体时,生成较多的1⁃甲丙基环己烷(收率为49.3%).S.T.Thompson等[74 ] 采用Re对Pd/C催化剂进行修饰,结果表明,在400 ℃原位还原后,PdRe/C可获得完全脱氧产物(苯和环己烷),其中苯的收率为35.0%.PdRe双金属催化剂结合了Pd/C的脱甲氧基和加氢功能与Re/C的对苯酚的脱氧功能. ...
... [73 ].H.Shafaghat等[73 ] 以Pd/C为催化剂,探究了醇类(甲醇、乙醇、1⁃丙醇、1⁃丁醇、异丙醇和2⁃丁醇等)替代H2 的可行性.结果表明,只有仲醇可以作为Pd/C上愈创木酚转化的H供体.同时,当使用异丙醇作为H供体时,生成较多的1⁃甲丙基环己烷(收率为49.3%).S.T.Thompson等[74 ] 采用Re对Pd/C催化剂进行修饰,结果表明,在400 ℃原位还原后,PdRe/C可获得完全脱氧产物(苯和环己烷),其中苯的收率为35.0%.PdRe双金属催化剂结合了Pd/C的脱甲氧基和加氢功能与Re/C的对苯酚的脱氧功能. ...
... [73 ]以Pd/C为催化剂,探究了醇类(甲醇、乙醇、1⁃丙醇、1⁃丁醇、异丙醇和2⁃丁醇等)替代H2 的可行性.结果表明,只有仲醇可以作为Pd/C上愈创木酚转化的H供体.同时,当使用异丙醇作为H供体时,生成较多的1⁃甲丙基环己烷(收率为49.3%).S.T.Thompson等[74 ] 采用Re对Pd/C催化剂进行修饰,结果表明,在400 ℃原位还原后,PdRe/C可获得完全脱氧产物(苯和环己烷),其中苯的收率为35.0%.PdRe双金属催化剂结合了Pd/C的脱甲氧基和加氢功能与Re/C的对苯酚的脱氧功能. ...
4
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
... 单金属Pd催化剂[73 ] 及PdNi[10 ] 、PdFe[53 ⁃54 ] 、PdRe[74 ] 双金属催化剂也被用于愈创木酚的加氢脱氧反应.基于Pd催化剂的产物种类也很多,有的以芳香族化合物为主[10 ,53 ,74 ] ,有的是芳环加氢饱和的产物[73 ] .H.Shafaghat等[73 ] 以Pd/C为催化剂,探究了醇类(甲醇、乙醇、1⁃丙醇、1⁃丁醇、异丙醇和2⁃丁醇等)替代H2 的可行性.结果表明,只有仲醇可以作为Pd/C上愈创木酚转化的H供体.同时,当使用异丙醇作为H供体时,生成较多的1⁃甲丙基环己烷(收率为49.3%).S.T.Thompson等[74 ] 采用Re对Pd/C催化剂进行修饰,结果表明,在400 ℃原位还原后,PdRe/C可获得完全脱氧产物(苯和环己烷),其中苯的收率为35.0%.PdRe双金属催化剂结合了Pd/C的脱甲氧基和加氢功能与Re/C的对苯酚的脱氧功能. ...
... ,74 ],有的是芳环加氢饱和的产物[73 ] .H.Shafaghat等[73 ] 以Pd/C为催化剂,探究了醇类(甲醇、乙醇、1⁃丙醇、1⁃丁醇、异丙醇和2⁃丁醇等)替代H2 的可行性.结果表明,只有仲醇可以作为Pd/C上愈创木酚转化的H供体.同时,当使用异丙醇作为H供体时,生成较多的1⁃甲丙基环己烷(收率为49.3%).S.T.Thompson等[74 ] 采用Re对Pd/C催化剂进行修饰,结果表明,在400 ℃原位还原后,PdRe/C可获得完全脱氧产物(苯和环己烷),其中苯的收率为35.0%.PdRe双金属催化剂结合了Pd/C的脱甲氧基和加氢功能与Re/C的对苯酚的脱氧功能. ...
... [74 ]采用Re对Pd/C催化剂进行修饰,结果表明,在400 ℃原位还原后,PdRe/C可获得完全脱氧产物(苯和环己烷),其中苯的收率为35.0%.PdRe双金属催化剂结合了Pd/C的脱甲氧基和加氢功能与Re/C的对苯酚的脱氧功能. ...
2
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
... 此外,还有一些科研工作者致力于Re[75 ] 、Rh[76 ] 、Ag[77 ] 和Au[78 ] 等贵金属催化剂用于愈创木酚加氢脱氧反应的研究.例如,J.B.Mao等[78 ] 在300 ℃和6.5 MPa H2 的条件下,利用Au/TiO2 ⁃A催化剂对愈创木酚加氢脱氧反应进行了探讨.结果表明,产物中酚类化合物的选择性为87.0%,TiO2 表面是脱除甲氧基的活化位点. ...
2
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
... 此外,还有一些科研工作者致力于Re[75 ] 、Rh[76 ] 、Ag[77 ] 和Au[78 ] 等贵金属催化剂用于愈创木酚加氢脱氧反应的研究.例如,J.B.Mao等[78 ] 在300 ℃和6.5 MPa H2 的条件下,利用Au/TiO2 ⁃A催化剂对愈创木酚加氢脱氧反应进行了探讨.结果表明,产物中酚类化合物的选择性为87.0%,TiO2 表面是脱除甲氧基的活化位点. ...
2
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
... 此外,还有一些科研工作者致力于Re[75 ] 、Rh[76 ] 、Ag[77 ] 和Au[78 ] 等贵金属催化剂用于愈创木酚加氢脱氧反应的研究.例如,J.B.Mao等[78 ] 在300 ℃和6.5 MPa H2 的条件下,利用Au/TiO2 ⁃A催化剂对愈创木酚加氢脱氧反应进行了探讨.结果表明,产物中酚类化合物的选择性为87.0%,TiO2 表面是脱除甲氧基的活化位点. ...
3
... 收率/%
文献 T /°Ct /hp (H2 )/MPa溶剂 1 Pt/Hβ H2 /500 ℃ 250 2 4.00 正十烷 98.0 环己烷(44.0) [69 ] 2 Pt/Hβ H2 /300 ℃ 350 2 0.10 100.0 芳香族化合物(85.0) [70 ] 3 Pt/Nb2 O5 H2 /300 ℃ 300 0.10 35.5 苯酚(19.1) [71 ] 4 Pt⁃Mo/TiO2 H2 /350 ℃ 285 1 4.00 94.0 环己烷(73.4) [72 ] 5a Pd/C 230 4 异丙醇 98.5 1⁃甲丙基环己烷(49.3) [73 ] 6 Ni@Pd/SD H2 /450 ℃ 450 1 0.10 18.0 苯酚(12.0) [10 ] 7 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 1 0.10 100.0 苯酚(50.0) [53 ] 8 Pd⁃Fe/Al⁃MCM⁃41 H2 /450 ℃ 400 0.10 98.0 含一个O产物(80.0) [54 ] 9 PdRe/C H2 /400 ℃ 300 0.10 90.0 苯(35.0) [74 ] 10 Re/TiO2 H2 /300 ℃ 280 3 2.00 正庚烷 100.0 环己烷+苯(92.1) [75 ] 11 Rh/ZrO2 H2 /350 ℃ 300 4 7.00 十二烷 100.0 环己烷(87.7) [76 ] 12 Ag/TiO2 ⁃A H2/ Ar/400 ℃ 300 3 3.00 正庚烷 77.0 苯酚(43.0) [77 ] 13 Au/TiO2 ⁃A 300 6.50 甲苯 43.1 酚类产物(37.5) [78 ] 注: a 异丙醇作为H供体. ...
... 此外,还有一些科研工作者致力于Re[75 ] 、Rh[76 ] 、Ag[77 ] 和Au[78 ] 等贵金属催化剂用于愈创木酚加氢脱氧反应的研究.例如,J.B.Mao等[78 ] 在300 ℃和6.5 MPa H2 的条件下,利用Au/TiO2 ⁃A催化剂对愈创木酚加氢脱氧反应进行了探讨.结果表明,产物中酚类化合物的选择性为87.0%,TiO2 表面是脱除甲氧基的活化位点. ...
... [78 ]在300 ℃和6.5 MPa H2 的条件下,利用Au/TiO2 ⁃A催化剂对愈创木酚加氢脱氧反应进行了探讨.结果表明,产物中酚类化合物的选择性为87.0%,TiO2 表面是脱除甲氧基的活化位点. ...




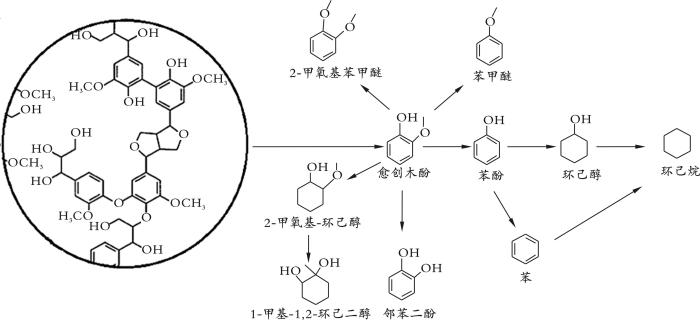


{kind=link}
{kind=link}